Control de calidad, alineación y llamada SNP.
La secuenciación ddRAD basó el genotipado de 58 individuos pertenecientes a seis razas bovinas nativas; Ganado Gir, Sahiwal, Tharparkar, Rathi, Red Sindhi y Kankrej con su distribución geográfica y ecológica (Fig. 1) incluido el propósito productivo, el color del pelaje, la zona agroclimática representativa, la zona de reproducción, la coordenada geográfica de cada zona de reproducción junto con la identificación del animal y el sexo de cada individuo presentado en la Tabla complementaria S1; resultó en 138.59 millones de lecturas sin procesar que correspondieron a 23 millones de lecturas por raza y 2.2 millones de lecturas por animal. Después del filtrado inicial según la calidad de lectura y la eliminación del adaptador, se retuvo la mayoría de las lecturas (138.58 millones de lecturas; 99.9%) (Tabla complementaria S2). Un alto porcentaje de lecturas (94.53%) se asignaron al Bos taurus (ARS-UCD1.2) conjunto de referencia (Tabla complementaria S2). En este estudio, se hizo el esfuerzo de analizar solo los SNP en diferentes razas de ganado, por lo que no se consideraron todas las demás variantes en análisis posteriores. El número de SNP en 6 razas de ganado osciló entre 8,42,768 y 3,81,966 después de la llamada de variante individual. El número máximo de SNP se observó en SAC (8,42,768), seguido de GIC (8,34,780), KAC (8,10,279), RAC (8,05,020), RSC (6,72,632) y THC (3,81,966). (Mesa 1). Los datos combinados establecidos en 6 razas de ganado produjeron un total de 43,47,445 SNP. Posteriormente, el archivo VCF se procesó paso a paso para filtrar los SNP de baja calidad. En primer lugar, los SNP se filtraron a una profundidad de lectura de 2 (RD 2), una profundidad de lectura de 5 (RD 5) y una profundidad de lectura de 10 (RD 10). Para un análisis más detallado, se utilizó el conjunto de datos de 9,82,174 SNP identificados en RD de 5 para el análisis posterior (Tabla 1). Todos los SNP que estaban presentes con baja cobertura (RD <5) se eliminaron del conjunto de datos. Los SNP que se identificaron con una RD de 5 se filtraron adicionalmente utilizando varios criterios, como la proporción de genotipos faltantes, la frecuencia de alelos menores y el equilibrio de Hardy Weinberg (HWE). La serie de filtrado dio como resultado un total de 84,027 SNP de alta calidad. Después del filtrado, la cantidad de SNP entre razas varió considerablemente. El mayor número de SNP se observó en GIC (34,743), seguido de RSC (13,092), KAC (12,812), SAC (8956), THC (7356) y RAC (7068) (Tabla 2).
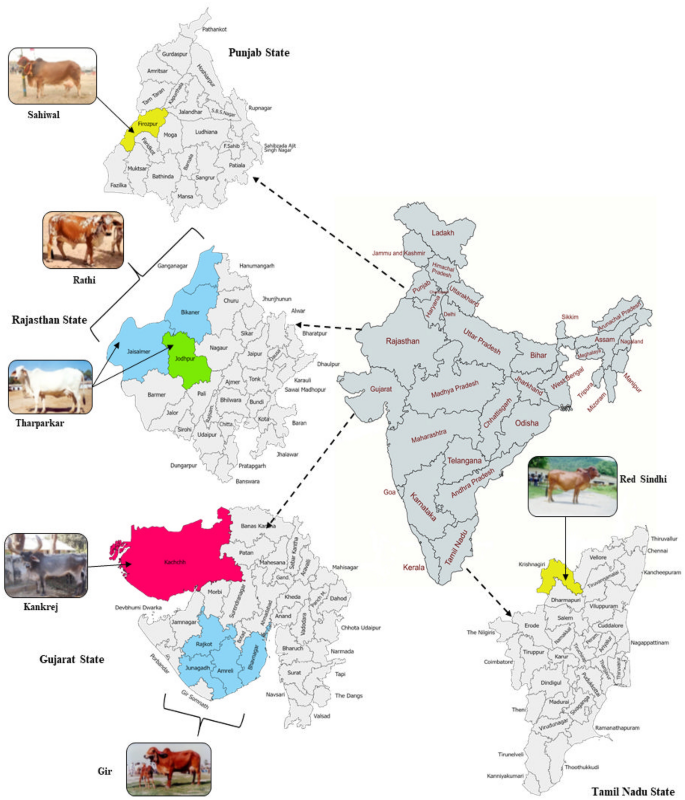
Distribución geográfica de seis razas de ganado incluidas en este estudio (El mapa se generó utilizando sitios web Map Chart https://www.mapchart.net/ y pintar mapas https://paintmaps.com/).
Anotación funcional de variantes.
El conjunto de datos combinados de SNP de alta calidad de las 6 razas lecheras se anotó para Bos taurus (ARS-UCD1.2) genoma de referencia. Con respecto a su distribución en el genoma, se predijo que una gran cantidad de SNP anotados estarían en la región intrónica (41,372 SNP, 53.87%) seguida de regiones intergénicas (26,834 SNP, 34.94%). Solo había 948 SNP (1.23%) distribuidos en las regiones exónicas. Además, había 3497 SNP (4.55%) ubicados dentro de la región de 5 Kb en sentido ascendente y 3661 SNP (4.77%) en sentido descendente del sitio de inicio de la transcripción. El análisis también dio como resultado 93 SNP (0.121 %) ubicados en 5'UTR, 293 SNP (0.38 %) en la región 3'UTR. También se predijo que un total de 8 SNP (0.01%) causarían codones de parada prematuros (Fig. 2).
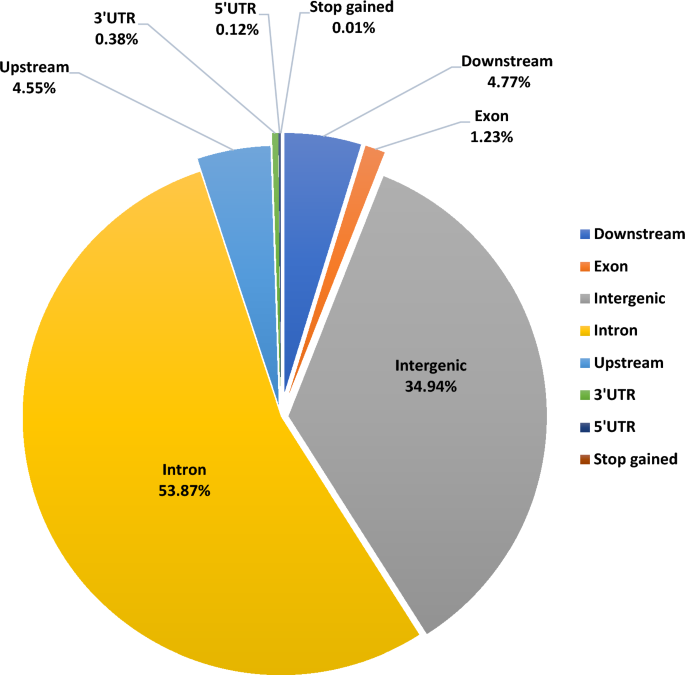
Partición general de SNP con respecto a la distribución genómica para todas las razas.
Sobre la base del impacto de los SNP en los genes codificantes de proteínas, los SNP se clasificaron como de alto impacto (10 SNP; 0.01%), impacto moderado (298 SNP; 0.39%) y bajo impacto (697 SNP; 0.91%). . La mayoría de los SNP (75,801; 98.69%) se identificaron como modificadores (Tabla complementaria S3). Además, una alta proporción de SNP (65.74 %) eran de naturaleza silenciosa, seguidos por los sin sentido (33.37 %) y los sin sentido (0.89 %), con una relación promedio sin sentido/silencioso de 0.507 (Tabla complementaria S4). Además, entre todos los genotipos sustituidos identificados en el presente estudio, se encontró que los genotipos C/T y G/A eran predominantes, mientras que el genotipo A/T se encontraba en las proporciones más bajas (Tabla complementaria S5). Para cada raza individual, los resultados de la anotación se resumen en la Fig. 3 y tabla suplementaria S6. En GIC, se predijo que el mayor número de SNP 32,283 (53.96%) estarían en la región intrónica seguida de la región intergénica 20,395 (34.09%). Sólo 777 (1.3%) fueron detectados en la región exónica. Al igual que en GIC, el mayor número de SNP se distribuyó en la región intrónica seguida de la región intergénica y exónica en todas las demás razas de ganado. Por ejemplo, en SAC, el 53.87% de los SNP (8429) se predijeron en la región intrónica, seguido del 33% en la región intergénica (5163 SNP) y solo el 1.75% (273 SNP) en la región exónica. Una tendencia similar se observó para las razas bovinas RAC, RSC, KAC y THC con 6834 (55.63%), 11,147 (52.12%), 8429 (53.87%), 6374 (52.58%) SNP, respectivamente en la región intrónica, 4186 (34.08%). %), 8192 (38.30%), 5163 (33%), 4507 (37.18%) SNP respectivamente, en la región intergénica y sólo 142 (1.16%), 266 (1.24%), 273 (1.75%), 123 (1.02 %) se predijeron en las regiones exónicas (Fig. 3). El número de variantes sinónimas identificadas en GIC, KAC, RAC, RSC, SAC y THC fue 570, 190, 101, 172, 213 y 87 respectivamente. Por otro lado, el número de variantes no sinónimas detectadas para las 6 razas bovinas fue 165, 64, 31, 82, 53 y 30 respectivamente. La tS/TV La proporción observada en GIC, KAC, RAC RSC SAC y THC fue de 2.55, 2.64, 2.33, 2.43, 2.51 y 2.19 respectivamente (Tabla complementaria S6).
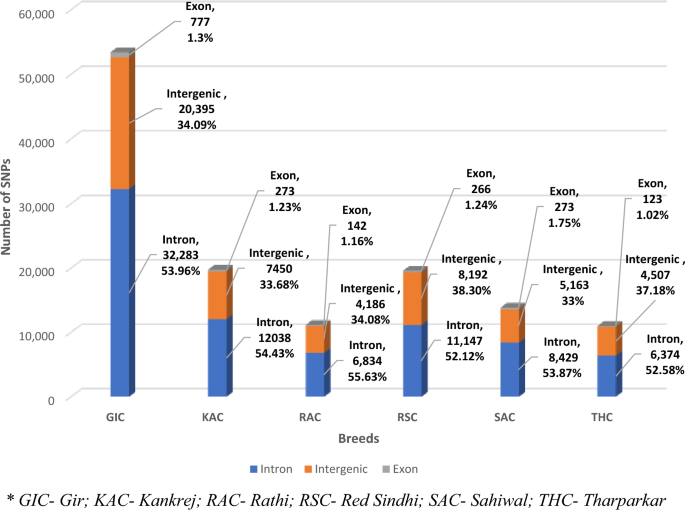
Distribución genómica de SNP en el genoma de seis razas de ganado lechero de la India.
El número de SNP intergénicos fue 4,639,873 (68.1%) y 1,676,710 (24.6%) eran intrónicos. Había 230,365 (3.4%) SNP ubicados dentro de 5 kb aguas arriba y 197,827 (2.9%) aguas abajo de un sitio de inicio de la transcripción; Se ubicaron 12,428 SNP en la UTR 5 ′ y 2613 en la UTR 3 ′. Un total de 4356 SNP se ubicaron en sitios de empalme de 2966 genes: 142 estaban en sitios donantes de empalme, 142 eran sitios receptores de empalme y 4072 estaban dentro de la región del sitio de empalme. Identificamos 45,776 SNP que afectan las secuencias codificantes de 11,538 genes. Se predijo que 221 SNP provocarían un codón de parada prematuro y 17 provocarían una ganancia en la secuencia codificante. El número de SNP que se predijo que no serían sinónimos fue 20,828. El número de SNP intergénicos fue 4,639,873 (68.1%) y 1,676,710 (24.6%) eran intrónicos. Había 230,365 (3.4%) SNP ubicados dentro de 5 kb aguas arriba y 197,827 (2.9%) aguas abajo de un sitio de inicio de la transcripción; Se ubicaron 12,428 SNP en la UTR 5 ′ y 2613 en la UTR 3 ′. Un total de 4356 SNP se ubicaron en sitios de empalme de 2966 genes: 142 estaban en sitios donantes de empalme, 142 eran sitios receptores de empalme y 4072 estaban dentro de la región del sitio de empalme. El número de SNP intergénicos fue 4,639,873 (68.1%) y 1,676,710 (24.6%) eran intrónicos. Había 230,365 (3.4%) SNP ubicados dentro de 5 kb aguas arriba y 197,827 (2.9%) aguas abajo de un sitio de inicio de la transcripción; Se ubicaron 12,428 SNP en la UTR 5 ′ y 2613 en la UTR 3 ′. Un total de 4356 SNP se ubicaron en sitios de empalme de 2966 genes: 142 estaban en sitios donantes de empalme, 142 eran sitios receptores de empalme y 4072 estaban dentro de la región del sitio de empalme. Identificamos 45,776 SNP que afectan las secuencias codificantes de 11,538 genes. Se predijo que 221 SNP provocarían un codón de parada prematuro y 17 provocarían una ganancia en la secuencia codificante. El número de SNP que se predijo que no serían sinónimos fue 20,828. El número de SNP intergénicos fue 4,639,873 (68.1%) y 1,676,710 (24.6%) eran intrónicos. Había 230,365 (3.4%) SNP ubicados dentro de 5 kb aguas arriba y 197,827 (2.9%) aguas abajo de un sitio de inicio de la transcripción; Se ubicaron 12,428 SNP en la UTR 5 ′ y 2613 en la UTR 3 ′. Un total de 4,356 SNP se ubicaron en sitios de empalme de 2966 genes: 142 estaban en sitios donantes de empalme, 142 eran sitios receptores de empalme y 4072 estaban dentro de la región del sitio de empalme. Identificamos 45,776 SNP que afectan las secuencias codificantes de 11,538 genes. Se predijo que 221 SNP provocarían un codón de parada prematuro y 17 provocarían una ganancia en la secuencia codificante. El número de SNP que se predijo que no serían sinónimos fue 20,828.
Dentro de la diversidad racial
La diversidad de nucleótidos (π) fue mayor en THC (π = 0.458), seguido de RSC (π = 0.364), SAC (π = 0.363), GIC (π = 0.356), KAC (π = 0.348) y RAC (π = 0.347). ). El valor medio de diversidad de nucleótidos fue 0.373 (Tabla 3). Los valores de D de Tajima fueron negativos para 4 razas de ganado a saber, RSC, RAC, SAC y THC, excepto para GIC y SAC donde se observaron valores de D positivos. El valor D negativo más alto de Tajima se observó en THC (-1.194), seguido de RSC (- 1.088), RAC (- 0.295) y KAC (- 0.279).
La heterocigosidad observada (HO) los valores oscilaron entre 0.464 y 0.551, mientras que la heterocigosidad esperada (HE) osciló entre 0.448 y 0.535. Los valores de heterocigosidad más altos observados se observaron en THC (HO = 0.551) seguido de RAC (HO = 0.523), RSC (HO = 0.5184), SAC (HO = 0.5180), GIC (HO = 0.499) y KAC (HO = 0.464) (Tabla 4). El promedio FIS (coeficiente de consanguinidad) oscila entre -0.253 en THC y 0.0513 en KAC. La FIS La estimación entre las seis razas de ganado fue la más alta en THC (FIS = − 0.253) seguido de RAC (FIS = − 0.105), mientras que el F más bajoIS estimación se observó en KAC (FIS = 0.0513) seguido de GIC (FIS = − 0.00063). La F generalIS El análisis reveló un exceso de heterocigosidad para todas las razas de ganado excepto KAC (Tabla 4). La heterocigosidad y FIS Las estimaciones indicaron la presencia de suficiente diversidad dentro de las seis razas de ganado.
Entre la diversidad racial
La diferenciación genética sobre la base del índice de fijación (FST) osciló entre 0.2840 y 0.3905, lo que indica suficiente diversidad entre razas. La mayor divergencia se observó entre el par RAC-SAC (FST = 0.3905), seguido del par racial RSC-RAC (FST = 0.3790), pareja de razas RSC-SAC (FST = 0.3751). La menor divergencia se observó para la pareja de razas KAC-THC (FST = 0.2840) (Tabla 5). El árbol basado en Neighbor Joining (NJ) construido agrupó los animales individuales de 6 razas de ganado según sus afiliaciones raciales, siendo GIC y RSC la raza más diversa entre las 6 razas de ganado estudiadas. La relación filogenética a nivel individual se muestra en la Fig. 4. El árbol de Nueva Jersey en cuanto a raza se muestra en la Fig. 5, más o menos corroborado con el árbol de niveles individuales. Además, se construyó un árbol filogenético basado en UPGMA a nivel de raza utilizando el paquete "phangorn" en la plataforma R con 100 valores de arranque. Los valores de arranque de cada nodo estuvieron cerca del 100%, lo que indica una alta robustez del árbol construido. El árbol filogenético basado en UPGMA refleja una relación genética similar a la revelada por la diferenciación genética basada en NJ (individualmente y a nivel de raza) donde GIC y RSC aparecieron como las razas más distintas. GIC apareció en el nodo principal y se agruparon como un grupo, mientras que las otras poblaciones formaron dos grupos con RSC agrupados en un nodo y RAC, THC, SAC y KAC formaron otros subgrupos (Fig. 6).
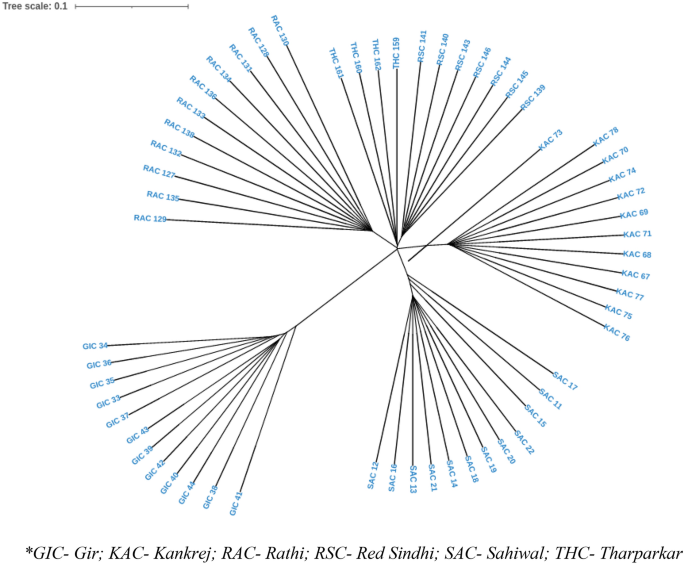
Agrupación filogenética basada en la unión de vecinos de 58 animales de seis razas de ganado lechero de la India utilizando el software Tassel.
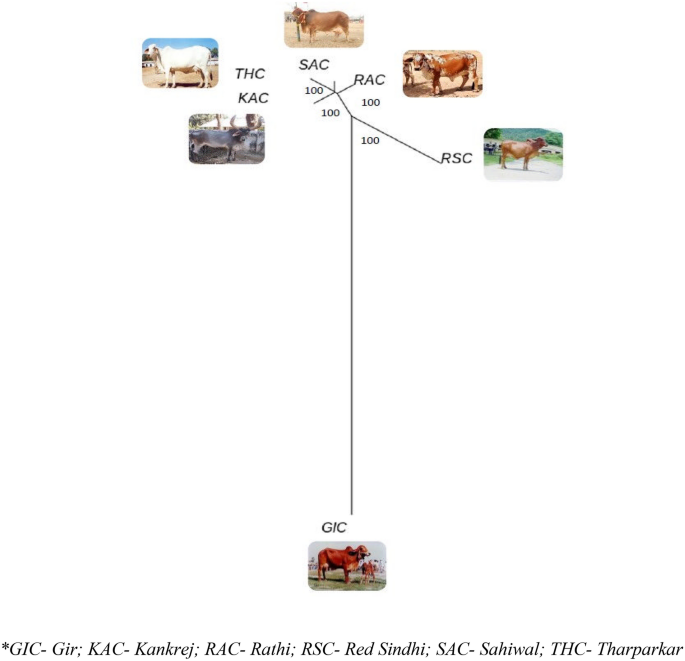
Agrupación basada en la unión de vecinos de 6 razas de ganado lechero de la India utilizando el paquete "phangorn" de la plataforma R.
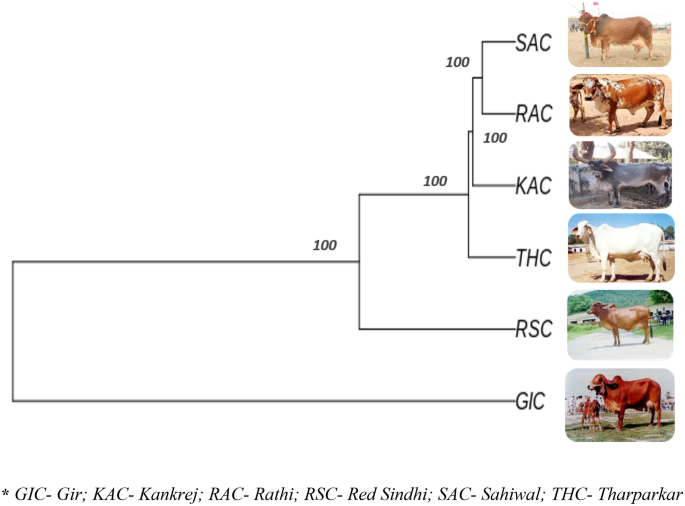
Agrupación filogenética basada en UPGMA de seis razas lecheras indias utilizando el paquete "phangorn" de la plataforma R.
Análisis de la estructura de la población.
El análisis de mezcla se llevó a cabo dividiendo el genoma de cada individuo en un grupo predefinido. El análisis se realizó a K = 3, 4, 5 y 6 (Fig. 7). Los individuos no pudieron agruparse en K = 3 según su raza respectiva. Sólo GIC podría diferenciarse claramente, mientras que los individuos de KAC y SAC aparecen como un grupo y RAC, THC y RSC están agrupados, lo que indica su ascendencia compartida. En K = 4, e incluso en K = 5, THC, RAC y RSC se agruparon indicando su fuerte ascendencia compartida, mientras que todos los demás individuos se agruparon en su propia raza respectiva. La mejor K en el análisis de la estructura poblacional es K = 6, por lo que casi todos los animales se agruparon en su raza respectiva, lo que indica claramente su ascendencia separada, con la excepción de RSC y THC que todavía se agruparon. La cercanía genética entre RSC y THC podría revelarse mediante más estudios en profundidad y aumentando el número de muestras.
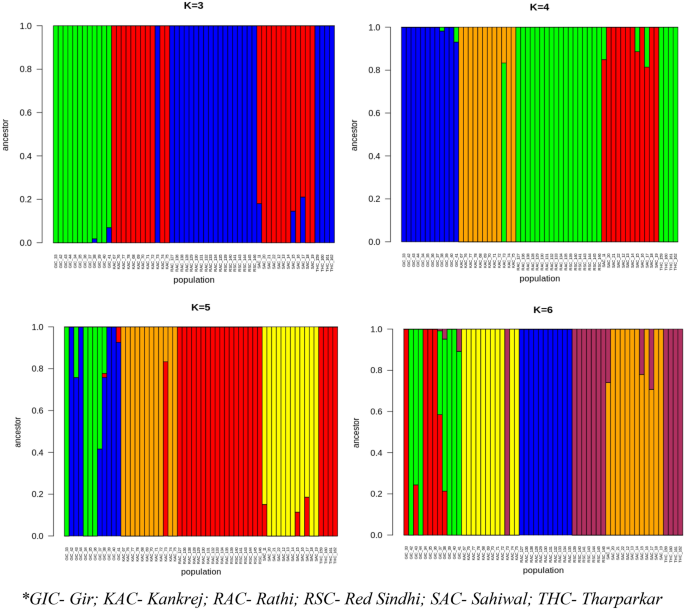
Análisis de mezcla suponiendo 3 ≤ K ≤ 6.
El análisis basado en PCA también agrupó 6 razas de ganado por separado y refuerza el hecho de que se trata de razas de ganado distintas (Figura complementaria. S1). Los individuos de KAC se agruparon en un cuadrante, mientras que los individuos de las razas bovinas SAC RAC, THC y RSC se ubicaron en un cuadrante diferente. Los individuos de la raza bovina GIC aparecieron como una población distinta.
- Distribución de relaciones públicas y contenido potenciado por SEO. Consiga amplificado hoy.
- EVM Finanzas. Interfaz unificada para finanzas descentralizadas. Accede Aquí.
- Grupo de medios cuánticos. IR/PR amplificado. Accede Aquí.
- PlatoAiStream. Inteligencia de datos Web3. Conocimiento amplificado. Accede Aquí.
- Fuente: https://www.nature.com/articles/s41598-023-32418-6

