La lesión muscular aguda induce la activación de Hedgehog, a través de su ligando DHH, para reprimir la formación de IMAT
La identidad del ligando Hh endógeno y su tipo de célula productora en el músculo esquelético sigue sin resolverse. Para determinar objetivamente qué ligando Hh se expresa por qué tipo de célula durante la regeneración muscular, utilizamos el análisis de expresión génica de una sola célula. Esta técnica supera las restricciones y los límites del método utilizado anteriormente, RT-qPCR de muestras de ARN a granel y, al mismo tiempo, permite visualizar la transcriptómica de todo el músculo a una resolución celular. Por lo tanto, extrajimos conjuntos de datos de RNAseq de una sola célula (scRNAseq) no publicados y públicos de músculo esquelético en diferentes puntos de tiempo antes y después de una lesión muscular aguda, como se describió anteriormente.41, para preguntar qué ligando de Hh se expresa en qué tipo de célula (Fig. 1a). Curiosamente, nuestros datos revelan que, Shh y Ihh son indetectables, mientras que Dhh se expresa potentemente dentro de las poblaciones endoteliales y neurales (principalmente compuestas por células de Schwann) (Fig. 1b). Nuestros datos también demuestran que Dhh se induce rápidamente tras la lesión antes de volver a los niveles de referencia a los 10 días posteriores a la lesión (dpi) (Fig. 1b). Estos datos confirman nuestras observaciones anteriores de que Dhh y no Shh es el ligando clave inducido por una lesión y expresado por las células de Schwann dentro del sistema nervioso periférico38,42. Curiosamente, también apuntan a las células endoteliales como otra fuente de DHH similar a lo que se ha informado después de las lesiones isquémicas.43.
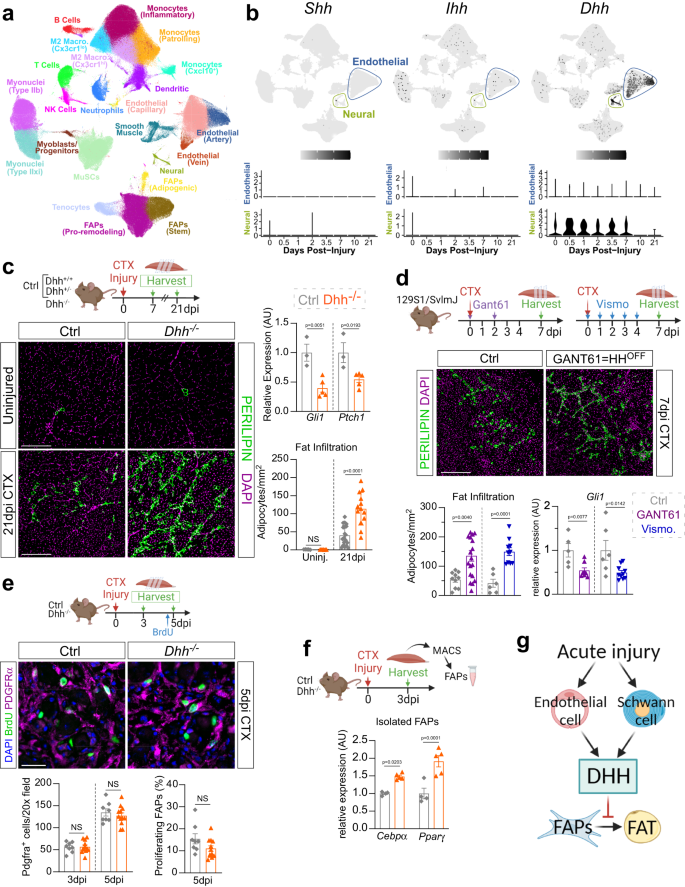
a Gráfico UMAP (Aproximación y proyección uniforme de la variedad) de 111 conjuntos de datos publicados de secuenciación de ARN de una sola célula y un solo núcleo del músculo esquelético antes y después de la lesión. b Gráficas UMAP que muestran la expresión logarítmicamente normalizada agregada de los ligandos Hh Shh (Sonic), Ihh (Indio)y Dhh (Desierto), y su expresión logarítmica normalizada dentro del grupo endotelial o neural en diferentes momentos posteriores a la lesión. c RT-qPCR para Gli1 y Ptch1 a 7 ppp (días posteriores a la lesión) posterior a la lesión por CTX (cardiotoxina) (ctrl (control) n = 3; Dhh- / - n = 5). Inmunofluorescencia para adipocitos (PERILIPIN+, verde) en el músculo TA (tibial anterior) lesionado y no lesionado. DAPI (púrpura) marca núcleos. Barra de escala: 250 µm. Cuantificaciones del número de adipocitos por área (Uninjured ctrl n = 5 Dhh- / - n = 7; Control de 21 ppp n = 20; Dhh- / - n = 13). d Inmunofluorescencia para PERILIPIN+ adipocitos (verde) a 7 dpi. DAPI (púrpura) marca núcleos. Barra de escala: 250 µm. Cuantificación de adipocitos por área lesionada Lesión CTX de 7 dpi de ratones administrados con Gant61 (vehículo tratado: n = 10; Gant61 tratado: n = 18) o Vismodegib (Vehículo ctrl: n = 6; Vismodegib tratado: n = 10). RT-qPCR para Gli1 de Gant61 (Vehículo ctrl: n = 5T; Gant61 tratado: n = 7) y Vismodegib (vehículo ctrl: n = 6; Vismodegib tratado: n = 10) ratones tratados. e Inmunofluorescencia de BrdU+ células (verde) y FAP (PDGFRα+ células; magenta) a 3 ppp. DAPI (púrpura) marca núcleos. Barra de escala: 25 µm. Cuantificaciones de (Unidades) PDGFRα total+ celdas por campo 20x 3 y 5 ppp en Dhh- / - (3 ppp n = 12; 5 ppp n = 12) y ctrl (3 ppp n = 8; 5 ppp n = 8) ratones. (Derecha) Porcentaje de FAP en proliferación (BrdU+ PDGFRα+ células; % del total de FAP) 5 dpi en Dhh- / - (n = 12) y ctrls (n = 8). f RT-qPCR para Cebpα y Pparγ de FAP purificados con MACS 3 dpi en Dhh- / - (n = 5 ratones) y ctrls (n = 4 ratones). g Modelo: Después de una lesión aguda, las células endoteliales y de Schwann expresan Dhh. La activación de Hh mediada por DHH reprime la formación de IMAT mediante la inhibición de la diferenciación adipogénica de FAP. Todos los datos se representan como media ± SEM. Cada punto de datos del gráfico en ce representa un músculo TA. Se utilizó una prueba t de dos colas para datos no apareados o un ANOVA unidireccional seguido de una comparación múltiple de Dunnet.
Para determinar no solo la función endógena de la señalización de Hh en general, sino la de DHH específicamente, utilizamos un modelo murino de ratón nulo Dhh (Dhh- / -) para evaluar cualquier defecto durante la regeneración muscular. Dhh- / - los ratones son viables, fenotípicamente normales y tienen una vida útil normal38,44. Confirmando y ampliando estos informes, encontramos que un niño de 7 meses ileso Dhh- / - los ratones no muestran anomalías fenotípicas graves, incluidas diferencias en el peso corporal total o el peso del tibial anterior (TA) sin lesiones en comparación con los controles de la camada (Dhh+/− y Dhh+ / +) (Fig. 1a). Primero investigamos si DHH induce la activación de Hh después de una lesión muscular aguda. Seleccionamos la cardiotoxina (CTX) como nuestro modelo de lesión, ya que induce la actividad de Hh8 y provoca poca formación de grasa45,46,47. Control (Dhh+ / + y Dhh+/−) y Dhh- / - los ratones se lesionaron con CTX y 7 días después de la lesión (dpi). La RT-qPCR de lisado muscular completo confirmó la pérdida completa de Dhh expresión en Dhh- / - ratones (Fig. 1b). La falta de DHH resultó en una disminución de la actividad de Hh como se muestra en la expresión de los dos objetivos de Hh aguas abajo Gli1 y Ptch1 in Dhh- / - en comparación con los ratones control (Fig. 1c). Por lo tanto, la pérdida de DHH previene la activación endógena de la señalización de Hh después de una lesión aguda.
Para determinar si la falta de activación de Hh podría afectar la formación de IMAT, cuantificamos el número de PERILIPIN+ adipocitos en TA no lesionados y lesionados de Dhh- / - y ratones de control. No encontramos diferencias en la formación de IMAT en TA no lesionados entre control y Dhh- / - ratones demostrando que la pérdida de Dhh no causa IMAT ectópico en ausencia de lesión (Fig. 1c). Emocionantemente, hubo un aumento significativo en IMAT 21 días después de la lesión CTX en Dhh- / - ratones en comparación con los controles (Fig. 1c). Esta observación fue independiente del sexo, y no detectamos ninguna diferencia entre Dhh+ / + y Dhh+/− animales de control (Fig. 1c). Además del aumento de IMAT, la mayoría de las enfermedades neuromusculares también se ven afectadas por la fibrosis tisular generalizada.28,48,49. Por lo tanto, también preguntamos si la pérdida de Dhh afectaría la fibrosis inducida por lesiones. Para visualizar la matriz extracelular (ECM), teñimos el control y Dhh- / - ratones 21 días después de la lesión de CTX con picrosirius red, que identifica las fibras de colágeno11,50. La falta de diferencia en la cantidad de depósito de colágeno entre los genotipos indica que la pérdida de señalización de Hh no tiene efecto sobre la fibrosis (Fig. 1d). Juntos, estos resultados demuestran que DHH actúa como un potente freno adipogénico para limitar la formación de IMAT, pero no la fibrosis, durante la regeneración muscular.
Para corroborar nuestros hallazgos, también utilizamos un enfoque farmacológico para inhibir de forma aguda la activación de Hh en varios momentos posteriores a la lesión. La pequeña molécula Gant61 es un inhibidor selectivo de la transcripción mediada por Gli1 y Gli2.51,52, mientras que Vismodegib es un inhibidor selectivo de Smo aprobado por la FDA53. Por lo tanto, ambas moléculas pequeñas previenen la activación endógena de Hh aguas abajo de la activación del ligando. Lesionamos los músculos TA de ratones de tipo salvaje con CTX y administramos Gant61 a 0 y 2 dpi, mientras que a una cohorte separada se le administró Vismodegib diariamente de 0 a 4 dpi (Fig. 1d). La inhibición exitosa de la activación de Hh inducida por CTX después del tratamiento con Gant61 y Vismodegib se determinó mediante RT-qPCR para Gli1 (Higo. 1d). Como resultado de la señalización de Hh reprimida, el tratamiento con Gant61 y Vismodegib permitió la formación de IMAT ectópicos (Fig. 1d) similar a la observada después de la pérdida de DHH (Fig. 1c). Por lo tanto, la inducción aguda de la señalización de Hh es necesaria para limitar la formación de IMAT durante la regeneración muscular.
Para definir el mecanismo celular sobre cómo DHH reprime la formación de IMAT, nos enfocamos en FAP, el origen celular de IMAT. Control y Dhh- / - los ratones se lesionaron con CTX y los TA se recolectaron a 3 y 5 ppp. Se administró bromodesoxiuridina (BrdU) 2 h antes de la recolección para visualizar las FAP en proliferación (Fig. 1e). No encontramos diferencias en el número total de PDGFRα+ FAP (Fig. 1e), ni hubo diferencia en la proliferación de FAP (Fig. 1e) que indica que no se requiere DHH para la expansión de FAP. Luego, determinamos si DHH podría afectar la diferenciación de FAP. Para ello, purificamos PDGFRα+ FAP utilizando la clasificación de células activadas magnéticamente (MACS) 3 días después de una lesión de CTX y evaluó la expresión de los genes adipogénicos tempranos clave Cebpα (proteína alfa de unión al potenciador de CCAAT) y Pparγ (Receptor gamma activado por el proliferador de peroxisomas) (Fig. 1f). Ambos genes adipogénicos fueron más fuertemente inducidos en PAF de Dhh- / - ratones en comparación con los controles (Fig. 1f) demostrando que la pérdida de Dhh permite la diferenciación adipogénica de FAPs. En conjunto, una lesión muscular aguda induce la expresión de Dhh por las células endoteliales y de Schwann, que a su vez bloquean la diferenciación de las FAP en adipocitos (fig. 1g).
DHH es necesario para la regeneración de miofibras
Nosotros y otros hemos demostrado que la activación ectópica de la vía Hh es suficiente para promover la regeneración de miofibras8,37,54,55. Para explorar si la señalización de Hh endógena, a través de su ligando DHH, también es necesaria para la miogénesis regenerativa, evaluamos la regeneración de miofibras 21 días después de la lesión de CTX en Dhh- / - y ratones de control (Fig. 2a). Para esto, medimos el área transversal (CSA) de las miofibras, una métrica crítica para evaluar la capacidad de las miofibras para recuperarse de una lesión.46. Las miofibras se visualizaron tiñendo secciones de tejido para LAMININ, un componente de ECM, mientras que las fibras regeneradas se seleccionaron en función de la presencia de núcleos ubicados centralmente. Después de segmentar y medir las miofibras utilizando nuestro canal de segmentación de miofibras recientemente desarrollado46, las miofibras se codificaron con colores falsos según el tamaño de la fibra (Fig. 2a). Similar a IMAT, no encontramos diferencias en la CSA de miofibra en TA no lesionados entre Dhh- / - y ratones de control argumentando que DHH es prescindible para la miogénesis embrionaria y la homeostasis adulta (Fig. 2a). Por el contrario, el CSA de miofibra disminuyó significativamente 21 días después de la lesión de CTX en Dhh- / - ratones en comparación con los controles (Fig. 2a). Esta reducción en el CSA promedio se debe a un cambio en la distribución del tamaño de la fibra, donde Dhh- / - los ratones muestran un cambio de fibras más grandes a más pequeñas (Fig. 2a). Por lo tanto, la pérdida de Dhh perjudica la regeneración de miofibras. Como confirmación independiente, también tratamos ratones de tipo salvaje con el antagonista de Hh Gant61 (Fig. 2a) y encontró una disminución en la miofibra CSA 7 días después de la lesión CTX (Fig. 2b) similar a la pérdida de Dhh. Por lo tanto, la activación endógena de Hh, a través de DHH, es fundamental para la regeneración de miofibras después de una lesión aguda.
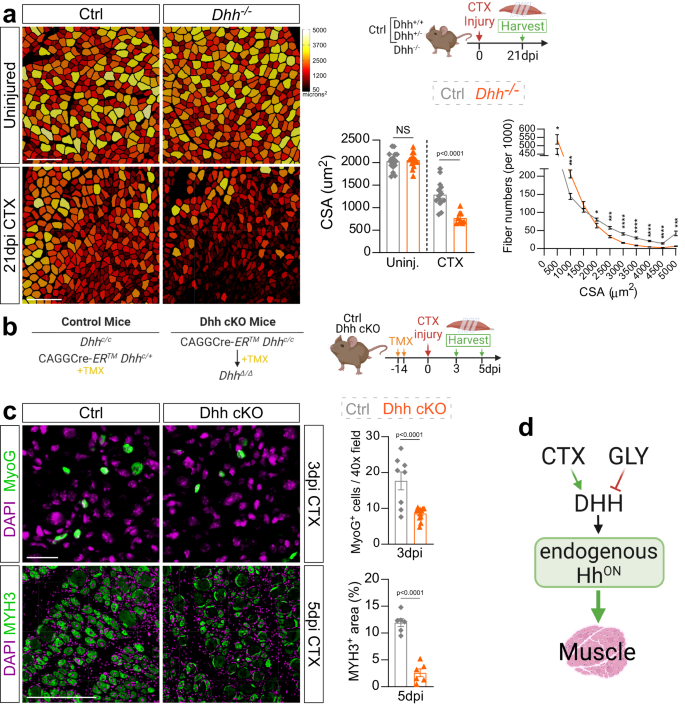
a Miofbras de ilesos y 21 días después de la lesión por CTX (cardiotoxina) de Dhh- / - y los ratones de control (ctrl) están codificados por colores según el área de la sección transversal (CSA; µm2). Barra de escala: 250 µm. Abajo a la derecha: CSA promedio (µm2) de ilesos (n = 16 TAs) y heridos (n = 9 TA) Dhh- / - en comparación con ilesos (n = 17 TAs) y heridos (n = 14 TAs) ratones control. Distribución del número de fibra basada en CSA de Dhh- / - (n = 9 TAs) y ratones ctrl (n = 14 TA) 21 dpi post CTX. *p = 0.03, ***p = 0.001 y ****p ≤ 0.0001. Los datos de origen se proporcionan como un archivo de datos de origen. b Esquema experimental. c Inmunofluorescencia y cuantificaciones de MYOG+ mioblastos (verde) en Dhh- / - (n = 11 TAs) y ratones ctrl (n = 8 TA) a 3 dpi (días posteriores a la lesión) después de CTX. Barra de escala: 25 µm. MI H+ miofibras (verde) en Dhh- / - (n = 6 TAs) y ratones ctrl (n = 6 TA) a 5 ppp post CTX. Barra de escala: 250 µm d Modelo: Se requiere DHH para la regeneración exitosa de miofibras. Los núcleos se visualizaron con DAPI (magenta). Todos los datos se representan como media ± SEM. Se utilizó una prueba t de dos colas para datos no apareados o un ANOVA unidireccional seguido de una comparación múltiple de Dunnet.
también nos lesionamos Dhh- / - ratones con glicerol (GLY), una lesión altamente adipogénica8,45,46,47,56,57. Primero, comparamos la activación de Hh entre ratones lesionados con CTX y GLY a través de RT-qPCR para Gli1 y Ptch1. Comparado con CTX, y similar a nuestros datos anteriores8, GLY no logra inducir la señalización de Hh (Fig. 2c). Lo importante es la pérdida de Dhh durante una lesión de GLY no disminuyó aún más la actividad de Hh (Fig. 2c). A continuación, analizamos el número de adipocitos que expresan PERILIPINA así como el tamaño de las miofibras 21 días después de la lesión por GLY y no encontramos diferencias entre Dhh- / - y ratones de control (Fig. 2d, e). Estos hallazgos indican que la pérdida de Dhh la lesión posterior a GLY no tiene impacto en los niveles de actividad de Hh, por lo que no causa ningún aumento adicional en IMAT. Por lo tanto, la activación diferencial de la señalización de Hh depende del tipo de lesión y los niveles de actividad de Hh dictan la cantidad de IMAT que se permite formar.
A continuación, buscamos explorar cómo Hh, a través de DHH, afecta los primeros pasos de la miogénesis regenerativa. Para ello, cruzamos un eliminador global Cre dependiente de tamoxifeno con un alelo Dhh floxed generado recientemente.43,58 (CAGGCre-ERTM xc / c, llamado Dhh cKO) (Fig. 2b). Este enfoque permite la pérdida generalizada y aguda de Dhh tras la administración de tamoxifeno justo antes de un insulto por lesión. Usando RT-qPCR, confirmamos la pérdida exitosa de Dhh expresión y reducción de la actividad de Hh (por Gli1 expresión) en ratones Dhh cKO 7 días después de la lesión por CTX en comparación con ratones compañeros de camada de control (Fig. 2f, gramo), Similar a Dhh- / - ratones (Fig. 1c). Es importante destacar que la eliminación inducible de Dhh condujo a una disminución en la miofibra CSA 7 días después de la lesión CTX (Fig. 2h) fenocopiando lo global Dhh fenotipo nulo (Fig. 2a). Para investigar el papel de DHH en la miogénesis temprana, después de la administración de TMX y el lavado, los TA se dañaron con CTX y se recolectaron a 3 y 5 ppp (Fig. 2b). A los 3 días después de la lesión por CTX, ya observamos una reducción dramática en los mioblastos que expresan MYOG (Fig. 2c). Oportunamente, esto resultó en una fuerte reducción en MYH3 recién formado+ (cadena pesada de miosina embrionaria 3) miofibras a 5 ppp. Por lo tanto, la pérdida de actividad de Hh da como resultado una reserva de mioblastos reducida, lo que perjudica la miogénesis temprana y la subsiguiente reparación de miofibras (Fig. 2d).
La activación ectópica de Hedgehog afecta tanto la adipogénesis como la miogénesis
Para determinar el momento en que se requiere la activación de Hh durante el proceso regenerativo, activamos farmacológicamente la vía de Hh en diferentes puntos de tiempo posteriores a la lesión de CTX o GLY mediante la administración del agonista suavizado, SAG59 (Higo. 3a). Primero evaluamos la cinética de SAG administrando una dosis única 2 días después de la lesión de GLY y evaluamos la inducción de Hh a través de la expresión de Gli1 y Ptch1 6 y 12 h después de la inyección (Fig. 3a). Curiosamente, encontramos que SAG induce rápidamente la señalización de Hh, lo que da como resultado una respuesta transcripcional dentro de las 6-12 h posteriores a la inyección (Fig. 3a). Para lograr una actividad Hh sostenida, administramos SAG en los días 0, 2 y 4 posteriores a la lesión. La activación temporal se llevó a cabo mediante una sola dosis de SAG el día de la lesión (0 dpi), 2 dpi o 4 dpi (Fig. 3a). Confirmamos la activación de Hh midiendo Gli1 expresión 7 días después de la lesión mediante RT-qPCR. Como era de esperar, observamos la mayor activación de Hh en ambos modelos de lesión cuando se administró SAG de forma repetitiva cada dos días (Fig. 3b) Mientras Gli1 los niveles volvieron a la línea de base el día 7 después de una dosis única de SAG a 0 dpi, la activación de Hh todavía estaba significativamente elevada cuando se administró SAG el día 4 con ambos modelos de lesión. Curiosamente, aún detectamos altas Gli1 los niveles de expresión 5 días después de que se administrara SAG 2 días después de la lesión por GLY, mientras que, por el contrario, la actividad de Hh ya no estaba elevada después de la lesión por CTX (Fig. 3b). Estos datos sugieren que el parámetro farmacocinético y/o dinámico de la actividad Hh inducida por SAG difiere entre los dos tipos de lesiones.
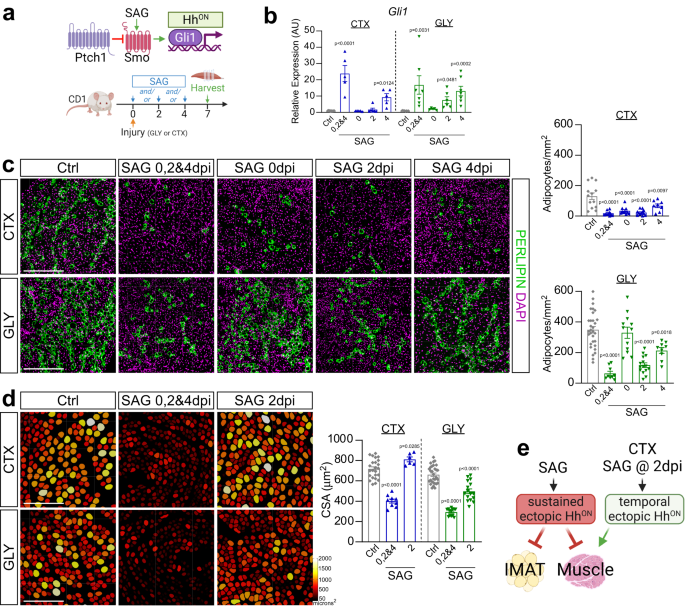
a Esquema experimental. b RT-qPCR para Gli1 7 días después de la lesión por CTX (cardiotoxina) o GLY (glicerol) cuando se trata con vehículo (CTX: n = 11; GLY: n = 8), SAG (agonista suavizado) a los 0, 2 y 4 días posteriores a la lesión (dpi) (CTX: n = 5; GLY: n = 7), a 0 ppp (CTX y GLY: n = 5), a 2 ppp (CTX y GLY: n = 6) o SAG a 4 ppp (CTX: n = 5; GLY: n = 7). c A la izquierda: Inmunofluorescencia para PERILIPIN+ adipocitos (verde) 7 días post CTX (parte superior) o GLY (fondo) lesión cuando se trata con vehículo, SAG a 0, 2 y 4 ppp, SAG a 0 ppp, SAG a 2 o 4 ppp. Los núcleos se visualizaron con DAPI (magenta). Barras de escala 250 µm. Derecha: Cuantificación de adipocitos por área lesionada (mm2) de CTX (parte superior) y GLY (fondo) 7 días después de la lesión. Control de vehículos (CTX: n = 13; GLY: n = 31), SAG a 0, 2 y 4 ppp (CTX: n = 10; GLY: n = 9), SAG a 0 ppp (CTX y GLY: n = 12), SAG a 2 ppp (CTX: n = 12; GLY: n = 15) o SAG a 4 ppp (CTX: n = 8; GLY: n = 9). d A la izquierda: Miofibras codificadas por colores basadas en el área de sección transversal (CSA) de CTX (parte superior) y GLY (fondo) ratones lesionados tratados con SAG a 0, 2 y 4 ppp, y SAG a 2 ppp. Barras de escala: 250 µm. A la derecha: CSA promedio (µm2) a 7 ppp. Control de vehículos (CTX: n = 27; GLY: n = 30), SAG a 0, 2 y 4 ppp (CTX: n = 9; GLY: n = 13), SAG a 2 ppp (CTX: n = 6; GLY: n = 17). e Modelo: La activación ectópica de Hh inducida por SAG bloquea la formación de IMAT en ambas lesiones. La activación ectópica sostenida de Hh también perjudica la regeneración muscular, mientras que un solo bolo de SAG en el día 2 después de CTX, pero no GLY, mejora la regeneración. Cada punto de datos del gráfico en bd representa un músculo TA. Todos los datos se representan como media ± SEM. Se utilizó ANOVA unidireccional seguido de una comparación múltiple de Dunnet.
A continuación, evaluamos los efectos de la activación de Hh sostenida versus temporal en la formación de IMAT a través de la cuantificación de PERILIPIN+ adipocitos. Confirmando nuestro trabajo previo y el de otros45,46,47, GLY, en comparación con CTX, indujo ~4 veces más IMAT (Fig. 3c). En el contexto de una lesión de CTX, la activación sostenida y temporal de Hh bloqueó significativamente la formación de IMAT, aunque en menor medida que cuando se activaba a 4 ppp (Fig. 3c). Por el contrario, la activación de Hh mostró una ventana de tiempo terapéutico más estrecha en el modelo GLY. Por ejemplo, IMAT posterior a la lesión de GLY se inhibió más fuertemente con la activación sostenida de Hh, seguida de la administración de SAG a los 2 ppp, con solo un impacto antiadipogénico modesto cuando se inyectó el día 4 (Fig. 3c). A diferencia de CTX, no hubo efecto sobre la formación de IMAT cuando se administró SAG en el momento de la lesión de GLY (Fig. 3c). Estos datos indican que la ventana de tiempo en la que los FAP son susceptibles a la represión adipogénica inducida por Hh es más amplia para una lesión de CTX con una ventana terapéutica muy estrecha entre 2 y 4 días después de la lesión para GLY.
Para determinar si la actividad de Hh también es necesaria para la regeneración de miofibras, evaluamos el tamaño de PHALLOIDIN+ miofibras para varios puntos de tiempo de dosificación de SAG 7 días después de la lesión de CTX y GLY (Fig. 3d). En ambos modelos de lesiones, la activación sostenida de Hh y la activación en el día 4 posterior a la lesión condujo a una disminución en el tamaño promedio de las miofibras, mientras que no se observó ningún efecto cuando se administró SAG el día de la lesión (Fig. 3d y Fig. Suplementaria. 3b). Inesperadamente, la activación de Hh a los 2 días de la lesión tuvo efectos opuestos según el tipo de lesión (fig. 3d). Similar a dos informes recientes54,55, la administración de SAG después de CTX promovió la miogénesis regenerativa (Fig. 3d) lo que indica que aumentar los niveles de Hh durante las lesiones agudas es beneficioso. Sin embargo, la administración de SAG 2 días después de la lesión por GLY perjudicó severamente la regeneración de las miofibras (Fig. 3d). Por lo tanto, la activación ectópica de Hh puede actuar como una señal regenerativa o degenerativa según el modelo de lesión y la ventana de tiempo (Fig. 3e).
Nuestros datos indican que una sola inyección de SAG a 2 ppp provoca una activación prolongada de Hh después de la lesión de GLY en comparación con CTX (Fig. 3b). Para determinar si los defectos de regeneración muscular que observamos en el modelo GLY se deben a una actividad Hh sostenida de alto nivel, administramos SAG a los 0, 2 y 4 días después de la lesión GLY a diferentes dosis: 2.5 mg/kg (1x dosis) se diluyó aún más a 1.25 mg/kg (dosis 0.5x); 0.83 mg/kg (0.3x dosis); o 0.5 mg/kg (dosis 0.2x) (Fig. 3c). Usando RT-qPCR, encontramos una inducción dependiente de la dosis de Gli1 expresión (Fig. 3c). Cuantificación del número de PERILPIN+ adipocitos, detectamos una fuerte correlación entre los niveles de Hh y la formación de IMAT (Fig. 3c). De manera similar, el tamaño de las miofibras, según las mediciones de CSA, también se inhibió de manera dependiente de la dosis (Fig. 3c). Por lo tanto, la activación ectópica de Hh posterior a la lesión de GLY bloquea IMAT pero también inhibe la regeneración de miofibras de una manera dependiente de la dosis.
Las FAP son el tipo de célula principal en el músculo esquelético que detecta y responde a DHH
Como Hh afecta tanto a la adipogénesis como a la miogénesis, a continuación queríamos identificar el tipo de célula a través del cual Hh ejecuta ambas funciones. Al preguntar qué tipos de células pueden ser capaces de inducir la señalización de Hh, primero extrajimos el mismo conjunto de datos de scRNAseq que se describió anteriormente para las células que expresan los factores de transcripción Gli. Gli1, Gli2 y Gli3. Descubrimos que las FAP y, en menor grado, las MuSC, son las dos poblaciones celulares principales que expresan las tres Glis en el músculo (Fig. 4a) y, por lo tanto, sería capaz de ejecutar la señalización Hh. Oportunamente, nosotros y otros hemos informado previamente que las FAP y las MuSC poseen un cilio primario8,55,60,61,62,63, un requisito previo para responder a Hh30.
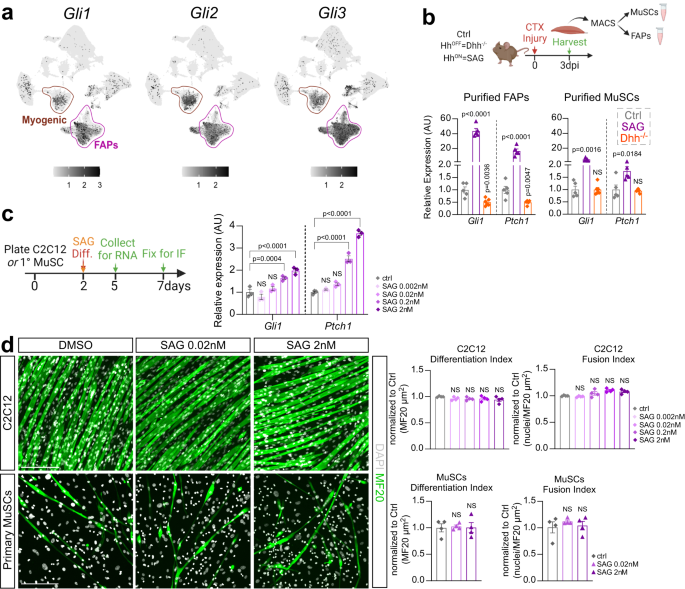
a Gráficas UMAP (Aproximación y proyección uniformes de la variedad) que muestran la expresión logarítmica agregada normalizada de los factores de transcripción Hh Gli1, Gli2 y Gli3. b RT-qPCR de expresión de Gli1 y Ptch1 a 3 dpi CTX (cardiotoxina) de MACS- (clasificación de células activadas magnéticamente) FAP purificadas aisladas (Unidades) y MuSC (Derecha) de control (ctrl) (n = 5), Dhh- / - (n = 5) y ratones tratados con SAG- (agonista suavizado) (n = 5). c Unidades: Diseño experimental. Derecha: RT-qPCR de expresión de Gli1 y Ptch1 de C2C12 (n = 3 repeticiones por grupo experimental) células 5 días después de la inducción de la diferenciación en ctrl y concentraciones variables de células tratadas con SAG (0.002 nM, 0.02 nM, 0.2 nM y 2 nM). d A la izquierda: Inmunofluorescencia de miofibras maduras marcadas con MF20 (cadena pesada de miosina; verde) y núcleos con DAPI (blanco) 7 días después de la inducción miogénica de C2C12 (parte superior) o MuSC primarias (fondo). Las células se trataron con vehículo (DMSO) o SAG (0.02 nM y 2 nM). Barras de escala: 200 µm. Derecha: Cuantificaciones (n = 4 repeticiones para todos los grupos experimentales) del índice de diferenciación (MF20+/μm2) e índice de fusión (núcleos por MF20+/μm2) de C2C12 (Notable ) o derivado primario de MuSC (Fondo) miofibras. Todos los datos se representan como media ± SEM. Se utilizó ANOVA de una vía seguido de una comparación múltiple de Dunnet.
A continuación, aislamos FAP y MuSC utilizando la clasificación de células activadas magnéticamente (MACS) de Dhh- / - y ratones tratados con SAG 3 días después de la lesión por CTX y compararon la inducción de los objetivos Hh Gli1 y Ptch1 como una lectura de la actividad de Hh a través de RT-qPCR (Fig. 4b). Como era de esperar, encontramos que la actividad de Hh se desactiva en los FAP tras la pérdida de Dhh y encendido en el tratamiento SAG. Sorprendentemente, solo detectamos una inducción débil de Hh en MuSC después del tratamiento con SAG, mientras que la expresión de ambos objetivos de Hh no cambió tras la pérdida de Dhh (Higo. 4b).
Para determinar si la débil activación de Hh en MuSC tiene algún impacto funcional en la regeneración de miofibras, realizamos un ensayo de miogénesis in vitro en presencia o ausencia de SAG usando C2C12 y MuSC primarias (Fig. 4c). Basado en RT-qPCR para genes diana Hh Gli1 y Ptch1, SAG indujo la señalización de Hh en células C2C12 a concentraciones crecientes. Sin embargo, de manera similar a nuestros resultados in vivo, la respuesta general de Hh fue débil. A continuación, determinamos el impacto de la actividad de Hh en la diferenciación de miofibras. La formación de miofibras se evaluó mediante la visualización de MF20+ fibras (índice de diferenciación), mientras que la tasa de fusión de mioblastos se calculó evaluando el número de mionúcleos (DAPI) dentro de MF20+ fibras (índice de fusión). Descubrimos que el tratamiento con SAG no afectó la diferenciación o la fusión de las células C2C12 o las MuSC primarias (Fig. 4d). Por lo tanto, nuestros datos demuestran que los FAP son los principales respondedores Hh celulares. Al mismo tiempo, mientras que las MuSC pueden responder a un estímulo Hh ectópico, aunque débilmente, esto no tiene impacto en su diferenciación en miofibras. Curiosamente, nuestros datos también destacan que las MuSC son insensibles al ligando Hh endógeno DHH.
La activación sostenida de Hedgehog afecta la diferenciación y supervivencia de FAP
Con base en nuestros resultados hasta el momento, la Hh debe afectar la miogénesis indirectamente a través de las FAP, el principal respondedor celular de la actividad de la Hh endógena (Fig. 4). Para probar genéticamente esta hipótesis, utilizamos un modelo de ratón condicional que, tras la administración de tamoxifeno (TMX), da como resultado la eliminación genética de Ptch1, un regulador negativo de la vía, dentro de las FAP (Pdgfrα-CreERT Ptch1c / c, llamado PAFsin PTCH1). Como informamos anteriormente8, pérdida de Ptch1 da como resultado la activación ectópica de Hh específica de FAP. Se administró tamoxifeno a ratones de 12 semanas de edad durante dos días consecutivos por sonda oral, seguido de un período de lavado de 2 semanas antes de lesionar los TA con GLY (Fig. 5a). Primero evaluamos la formación de IMAT cuantificando el número de adipocitos que expresan PERILIPINA (Fig. 5b). Si bien no hubo diferencia en la cantidad de IMAT en ausencia de lesión (Fig. 4a), la formación de IMAT se bloquea fuertemente después de GLY (Fig. 5b) y lesión CTX (Fig. 4b) en PAFsin PTCH1 ratones en comparación con los controles de compañeros de camada. Estos resultados confirman y amplían nuestros hallazgos previos de que la formación de IMAT permanece reprimida más allá de los 7 días, independientemente del tipo de lesión, tras la activación de Hh específica de FAP8.
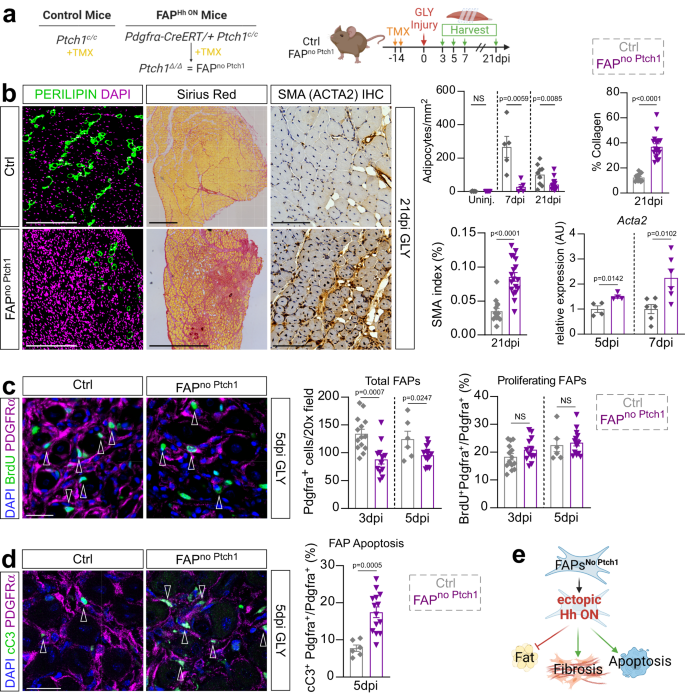
a Esquema experimental. b Unidades: Inmunofluorescencia para PERILIPIN+ adipocitos (verde) 21 días post GLY de ctrl y FAPsin Ptch1 ratones. DAPI (púrpura) marca núcleos. Barras de escala: 250 µm. Medio: Tinción histológica con rojo de Sirio. Barras de escala: 500 µm. A la derecha: Inmunohistoquímica para αSMA (actina de músculo liso) a 21 dpi (días posteriores a la lesión) glicerol (GLY). Barras de escala: 100 µm. Más a la derecha: Cuantificación de adipocitos de ilesos (FAPsin Ptch1 n = 5; control n = 5) ratones, 7 días (FAPsin Ptch1 n = 5; control n = 5) y 21 días post GLY (FAPsin Ptch1 n = 14; ctrls n = 9). Cuantificación del porcentaje de colágeno rojo Sirius positivo (rojo) del área total de TA de FAPsin Ptch1 (n = 18) y control (n = 12) ratones 21 ppp. Cuantificación del porcentaje de αSMA+ celdas (marrones) del área total de TA de FAPsin Ptch1 (n = 18) y control (n = 14) ratones 21 ppp. RT-qPCR de expresión de Acta2 a 5 ppp (n = 4 para ambos) y 7dpi (n = 6 para ambos). c Unidades: Inmunofluorescencia de BrdU+ células (verde) y FAP (PDGFRα+ células; magenta) 5 días después de la lesión por CTX (cardiotoxina) en ctrl y FAPsin Ptch1 ratones. Las puntas de flecha apuntan a celdas positivas dobles. DAPI (púrpura) marca núcleos. Barra de escala: 25 µm. Derecha: Cuantificaciones de FAP totales por campo 20x a 3dpi (FAPsin Ptch1 n = 12; control n = 14) y 5 ppp (FAPsin Ptch1 n = 14; control n = 6); y porcentaje de FAP en proliferación (BrdU+ Pdgfrα+ células; % del total de FAP) a 3dpi (FAPsin Ptch1 n = 12; control n = 14) y 5 ppp (FAPsin Ptch1 n = 14; control n = 6). d Unidades: Inmunofluorescencia de FAPs (PDGFRα+; magenta) experimentando apoptosis, marcada por Caspasa 3 escindida (cC3+; verde; puntas de flecha) 5 días después de GLY en ctrl y FAPsin Ptch1 ratones. DAPI (púrpura) marca núcleos. Barra de escala: 25 µm. Derecha: Cuantificaciones del porcentaje de FAP que experimentan apoptosis (% del total de FAP) a 5 ppp en FAPsin Ptch1 (n = 14) y ctrl (n = 6) ratones. e Modelo: La activación ectópica reprime la conversión adipogénica de las PAF, afecta su supervivencia y promueve la fibrosis. Cada punto de datos del gráfico en (b–d) representa un músculo TA. Todos los datos se representan como media ± SEM. Se utilizó una prueba t de dos colas para datos no apareados o un ANOVA unidireccional seguido de una comparación múltiple de Dunnet.
Además de los adipocitos, los FAP pueden diferenciarse en miofibroblastos, el origen celular del tejido cicatricial fibrótico.7,15. Dado que se desconoce el destino de las PAF que no se han diferenciado en adipocitos, preguntamos si la activación ectópica de Hh puede haber empujado a las PAF hacia un destino fibrótico. Para ello evaluamos la fibrosis 21 días después de una lesión de GLY y CTX mediante una tinción con rojo Sirius y encontramos un aumento significativo del depósito de colágeno en FAPsin Ptch1 en comparación con los controles (Fig. 5b y Fig. Suplementaria. 4b). A continuación, evaluamos si la fibrosis se debe a la diferenciación de FAPs hacia miofibroblastos. Apropiadamente, la tinción inmunohistoquímica (IHC) contra la actina del músculo liso α (Acta2, también llamada α-SMA), un marcador de miofibroblastos, 21 días después de una lesión GLY mostró un aumento dramático en las células que expresan α-SMA en el FAPsin Ptch1 ratones en comparación con los controles (Fig. 5b). Confirmando estos hallazgos, encontramos por RT-qPCR que ya había un aumento significativo en la expresión de Acta2 en PAFsin Ptch1 ratones en puntos de tiempo anteriores (Fig. 5b). Estos hallazgos indican que la activación ectópica de Hh puede empujar a los FAP a adoptar un destino de miofibroblastos.
Para definir si esta conversión fibrótica depende del momento de la activación de Hh, también cuantificamos la deposición de colágeno después de la activación de Hh temporal frente a la sostenida a través de SAG posterior a la lesión de CTX y GLY (Fig. 4c). Curiosamente, encontramos un aumento del rojo Sirius+ el depósito de colágeno 7 días después de la lesión después de la activación sostenida de Hh (SAG administrado a 0, 2 y 4 ppp), mientras que la activación temporal (SAG administrado solo a 2 ppp) no tuvo efecto, independientemente del modelo de lesión (Fig. 4c). Esto indica que solo la activación de Hh sostenida pero no temporal causa fibrosis.
Si bien la actividad endógena de Hh no tuvo impacto en la proliferación o el número total de FAP (Fig. 1e), queríamos dilucidar si la Hh ectópica puede estar influyendo en la proliferación y/o supervivencia de FAP. Curiosamente, encontramos que hay una disminución en el número total de PDGFRα+ células en PAFsin Ptch1 ratones en comparación con los controles 3 y 5 ppp después de la lesión por GLY (Fig. 5c). Sin embargo, no detectamos diferencias en la tasa de proliferación de FAP (BrdU+ PDGFRα+ células) entre genotipos (Fig. 5c). Para determinar si el aumento de la muerte celular podría explicar esta disminución en el número total de FAP, co-teñimos los FAP para el marcador de apoptosis escindido Caspase3 (cCas3). Curiosamente, vimos un aumento significativo de cCas3+ PDGFR-α+ FAP en FAPsin Ptch1 ratones en comparación con los controles a 5dpi (Fig. 5d) lo que indica que el aumento de la muerte celular es la explicación más probable para la reducción del número de PAF. Por lo tanto, la activación sostenida de Hh provoca un aumento de la muerte celular que da como resultado menos PAF. Proponemos que las FAP restantes se vean obligadas a adoptar un miofibroblasto en lugar de un destino adipogénico, lo que provoca la formación masiva de tejido cicatricial (Fig. 5e).
La activación de Hh específica de FAP afecta gravemente la miogénesis regenerativa
Al analizar la FAPsin PTCH1 ratones, notamos que los TA de FAPsin Ptch1 los ratones parecían más pequeños en tamaño en comparación con los controles (Fig. 5b). Tras un examen más detenido, mientras que los TA teñidos con H&E no lesionados tenían un tamaño comparable entre los genotipos (Fig. 5a), hubo una disminución significativa en el área total 21 días después de GLY en TA de FAPsin PTCH1 ratones (Fig. 6a). Para determinar si el cambio en el tamaño de TA se debe a la pérdida de miofibras, cuantificamos el número de miofibras presentes en las secciones transversales de TA en FAPsin PTCH1 y ratones de control como proxy del número total de fibras presentes por TA (Fig. 6a). No pudimos detectar ninguna diferencia en la cantidad de fibras musculares que indique que la regeneración de miofibras aún es funcional en FAPsin PTCH1 ratones (Fig. 6a). A continuación, preguntamos si las diferencias de tamaño de TA se deben a una regeneración de miofibras retrasada o incompleta que da como resultado miofibras más pequeñas al medir los CSA de miofibras individuales entre genotipos (Fig. 6a). No hubo diferencia en CSA sin lesión (Fig. 6a y Fig. Suplementaria. 5a), destacando que la activación de Hh específica de FAP no es necesaria para la homeostasis de las miofibras. En contraste, mientras que los ratones de control muestran una recuperación significativa de miofibras de 7 a 21 días después de la lesión GLY, FAPsin PTCH1 los ratones no lograron aumentar el tamaño de sus miofibras (Fig. 6a) y mostró un cambio dramático en el tamaño de las miofibras hacia fibras más pequeñas (Fig. 5b). Esto también fue cierto 21 días después de la lesión de CTX (Fig. 4c) lo que indica que el impacto en la miogénesis es independiente de la lesión. Cuantificamos aún más el contenido mionuclear de cada miofibra regenerada y descubrimos que FAPsin PTCH1 los ratones tenían significativamente menos mionúcleos por fibra que los controles (Fig. 6a). Esto sugiere que la activación ectópica de Hh específica de FAP perjudica la regeneración de miofibras de una manera celular no autónoma.
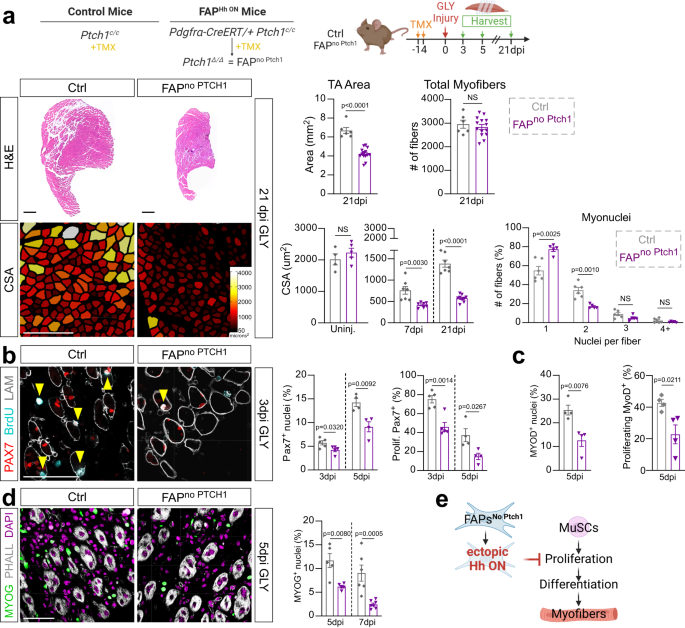
a Unidades: Tinción con hematoxilina y eosina (H&E) de FAPsin Ptch1 y ratones ctrl 21 dpi (días posteriores a la lesión). Barras de escala: 500 µm. Miofibras codificadas por colores según el tamaño de FAPsin Ptch1 y ratones ctrl 21 dpi. Barras de escala: 250 µm. Derecha: Cuantificación del área total de TA 21 días post GLY (glicerol) de FAPsin Ptch1 (n = 14) y ctrl (n = 6) ratones. Cuantificación de números de fibras totales 21 dpi de FAPsin Ptch1 (n = 14) y ctrl (n = 6) ratones. Promedio de CSA (área transversal) de personas ilesas (FAPsin Ptch1 n = 5; control n = 4), 7 ppp (FAPsin Ptch1 n = 7; control n = 7) y 21 ppp (FAPsin Ptch1 n = 9; control n = 7) ratones. Distribución del número de mionúcleos por fibra (% de fibras totales) de FAPsin Ptch1 (n = 5) y ctrl (n = 6) ratones 21 ppp. b A la izquierda: Inmunofluorescencia para PAX7+ (MuSCs (células madre musculares), rojo), BrdU+ (células en proliferación, cian) y LAMININA (contorno de miofibras, blanco) 3 dpi. Células positivas dobles marcadas con punta de flecha. Barra de escala: 50 µm. A la derecha: Cuantificaciones de porcentaje de PAX7+ núcleos (% del total de núcleos) y PAX7 en proliferación+ MuSCs (% del total de MuSCs) a 3 dpi (n = 5 para cada uno) y 5 ppp (n = 4 para cada uno). c Porcentaje de MYOD+ y BrdU+ MIO+ mioblastos a 5 dpi en FAPsin Ptch1 (n = 4) y ctrl (n = 4) ratones. d Unidades: Inmunofluorescencia de MYOG+ núcleos (verde) y FALLOIDINA (blanco) 5 dpi. Núcleos visualizados por DAPI (magenta). Barra de escala: 50 µm. Derecha: Porcentaje de MYOG+ núcleos a 5 ppp (n = 5 para cada uno) y 7 dpi (FAPsin Ptch1 n = 7 TA; control n = 6 TA). e Modelo: La activación ectópica de Hh en FAP reprime indirectamente la proliferación y expansión de MuSC, lo que da como resultado una reserva de mioblastos reducida y miofibras más pequeñas. Cada punto de datos del gráfico en el anuncio representa un músculo TA. Todos los datos se representan como media ± SEM. Se utilizó una prueba t de dos colas para datos no apareados o un ANOVA unidireccional seguido de una comparación múltiple de Dunnet.
Tras la lesión muscular, MuSCs (PAX7+ células) se activan, se expanden y hacen la transición a lo largo de un proceso gradual, controlado por factores de transcripción pro-miogénicos como MyoD y MyoG, para generar mioblastos. Estos mioblastos, a su vez, continúan proliferando antes de diferenciarse en miocitos y luego se fusionan para reparar las miofibras dañadas o reemplazarlas.3,64.Por lo tanto, tratamos FAPsin PTCH1 y ratones de control con BrdU y evaluaron cualquier diferencia en la miogénesis temprana. Curiosamente, encontramos que la activación de Hh dentro de los FAP condujo a una disminución en el número total de PAX7+ MuSC debido a la proliferación defectuosa de MuSC como es evidente por la frecuencia reducida de BrdU+ PAX7+ células en comparación con los controles (Fig. 6b). La activación de Hh específica de FAP ectópica también bloqueó la proliferación y expansión de MyoD+ mioblastos (fig. 6c) resultando en un MyoG más pequeño+ grupo de mioblastos (Fig. 6d). En conjunto, nuestros datos demuestran que la activación ectópica de Hh de alto nivel, indirectamente a través de FAP, suprime la expansión de MuSC, lo que da como resultado una reserva de mioblastos reducida y, en última instancia, conduce a miofibras más pequeñas (Fig. 6e).
DHH controla la adipogénesis a través de TIMP3, mientras que afecta la miogénesis indirectamente a través de GDF10 producido por FAP
Los FAP son cruciales durante la reparación muscular al secretar numerosos factores beneficiosos5. Nuestros datos establecen que los FAP son los principales respondedores de Hh y que el impacto de Hh tanto en la adipogénesis como en la miogénesis es a través de los FAP. Para determinar el mecanismo molecular aguas abajo de las FAP, realizamos una prueba de inmunoabsorción ligada a enzimas (ELISA) para detectar múltiples citocinas de lisados de proteínas musculares completas de control y FAP.sin Ptch1 ratones (Fig. 6a). No encontramos diferencias en >30 dianas entre genotipos, lo que indica que no hay grandes alteraciones dentro de la respuesta inmune cuando Hh se activa de manera específica para FAP. A continuación, evaluamos los niveles de expresión de objetivos FAP conocidos (Ccl2, Ccl7, Cxcl5, Fst, Gdf10, Igf1, Igf2, Il10, Il4, Il6, spp1, tim3 y Wisp1) que se han descrito para influir en la regeneración muscular y la deposición de IMAT tanto en FAPsin Ptch1 y Dhh- / - ratones (Fig. 6b). Identificamos 2 genes (factor de diferenciación de crecimiento 10; gdf10 e inhibidor tisular de metaloproteinasa 3; timp3) que estaban significativamente regulados al alza en FAPsin Ptch1 y regulado a la baja en Dhh- / - ratones (Fig. 6c). Además, confirmamos que los FAP están expresando de manera específica y diferencial gdf10 y timp3 aguas abajo de la vía Hh por PDGFRα aislador de MACS+ FAP 3 días después de la lesión de CTX del control, SAG tratado y Dhh- / - ratones (Fig. 7a). En comparación con los ratones de control, en ausencia de DHH, gdf10 y timp3 están significativamente regulados a la baja dentro de los FAP (Fig. 7a). Por el contrario, tras la activación ectópica de Hh a través de SAG, los FAP aumentan significativamente tanto gdf10 y timp3 (Higo. 7a). Así, gdf10 y timp3 son expresados por FAPs, y su expresión depende de los niveles de actividad de Hh.
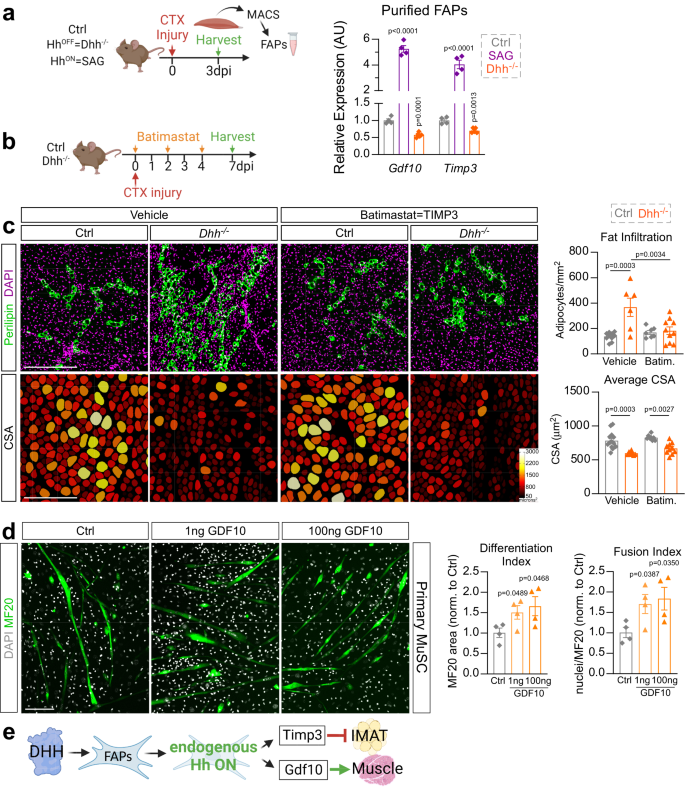
a RT-qPCR de gdf10 y timp3 dentro de MACS- (clasificación de células aisladas magnéticas) FAP aisladas (3 ppp (días posteriores a la lesión) CTX (cardiotoxina)) de ctrl (n = 4 ratones), Dhh- / - (n = 6 ratones) y ratones tratados con SAG (agonista suavizado) (a 0 y 2 ppp, n = 4 ratones). b Diseño experimental. c Unidades: Inmunofluorescencia para PERILIPIN+ adipocitos (verde) y DAPI+ núcleos (magenta). FALOIDINA+ miofibras (codificadas por colores según el área de la sección transversal (CSA)) 7 días después de la lesión por CTX del vehículo o tratado con batimastat Dhh- / - y ratones ctrl. Barras de escala: 250 µm. Derecha: Cuantificación del número de adipocitos por área lesionada de vehículo tratado Dhh- / - (n = 6) y ctrl (n = 10) en comparación con los tratados con batimastat Dhh- / - (n = 10) y ctrl (n = 7) ratones. CSA promedio (µm2) de miofibras 7 dpi de vehículo tratado Dhh- / - (n = 7) y ctrl (n = 14) en comparación con los tratados con batimastat Dhh- / - (n = 10) y ctrl (n = 8) ratones. Cada punto de datos del gráfico representa un músculo TA. d Unidades: Inmunofluorescencia de MF20+ (cadena pesada de miosina) miofibras (verde) diferenciadas de MuSC primarias (células madre musculares), 7 días después de la inducción y tratadas con GDF10 (1 ng y 100 ng) o vehículo de control (PBS). Los núcleos se visualizaron con DAPI (blanco). Barra de escala: 150 µm. Derecha: Cuantificaciones (n = 4 repeticiones para todos los grupos experimentales) para la diferenciación (MF20+/μm2) e índice de fusión (núcleos por MF20+/μm2). e Modelo: Después de una lesión aguda, DHH activa Hh en FAP, induciendo la expresión de Timp3 y Gdf10, que a su vez inhiben la adipogénesis o promueven la miogénesis, respectivamente. Todos los datos se representan como media ± SEM. Se utilizó un ANOVA unidireccional seguido de una comparación múltiple de Dunnet.
Anteriormente identificamos TIMP3 como un factor antiadipogénico dependiente de Hh específico de FAP8. En nuestro presente estudio, demostramos que la expresión endógena de timp3 también depende de la actividad de Hh. Para probar funcionalmente si la pérdida de actividad de TIMP3 en Dhh- / - ratones es la causa del aumento de la formación de IMAT, diseñamos un experimento de rescate en el que restauramos la función TIMP3 a través de la pequeña molécula Batimastat en Dhh- / - ratones. Batimastat es un inhibidor de panmetaloproteinasas que, como hemos demostrado anteriormente, puede actuar como un mimético farmacológico de TIMP3 e inhibe de forma potente la formación de IMAT.8. Se administró batimastat para controlar y Dhh- / - compañeros de camada, y a los 7 días después de la lesión por CTX se evaluó la formación de IMAT (Fig. 7b). Como era de esperar, vimos un aumento en PERIPIN+ adipocitos y una disminución de miofibras CSA en Dhh- / - en comparación con los ratones de control en el grupo tratado con vehículo (Fig. 7c). Emocionantemente, el tratamiento con Batimastat pudo bloquear PERILIPIN+ infiltración de adipocitos en Dhh- / - ratones. También se ha demostrado que Batimastat mejora la función muscular en mdx ratones, un modelo de ratón de distrofia muscular de Duchenne65. Por lo tanto, también evaluamos cualquier mejora en la regeneración de miofibras en pacientes tratados con Batimastat. Dhh- / - ratones. Sin embargo, no detectamos ninguna mejora en la CSA de miofibra en comparación con los controles tratados con Batimastat (Fig. 7c). Por lo tanto, DHH controla la formación de IMAT, pero no la regeneración de miofibras, a través de TIMP3.
GDF10 es un miembro de la superfamilia del factor de crecimiento transformante-β (TGFβ) y se ha identificado recientemente como un factor pro-miogénico10 secretada por FAP10,66. Por lo tanto, a continuación determinamos si GDF10 derivado de FAP controla la miogénesis. Primero confirmamos la activación transcripcional a largo plazo por GDF10 midiendo la expresión de Serpina 1 (también conocido como Pai1), un objetivo transcripcional conocido de GDF1067, 3 días después del tratamiento de células C2C12 con GDF10 recombinante (rGDF10) (Fig. 6d). A continuación, tratamos las MuSC primarias aisladas con rGDF10 durante la diferenciación miogénica (Fig. 7d). Después de 7 días en cultivo, calculamos los índices de diferenciación y fusión de miofibras mediante la visualización de miofibras maduras (MF20+ fibras) junto con los núcleos (DAPI) (Fig. 7d). Descubrimos que hubo un aumento tanto en la diferenciación como en la fusión de mioblastos cuando las MuSC primarias se trataron con proteína rGDF10 (Fig. 7d). Juntos, nuestros resultados indican que la activación endógena de Hh, a través de su ligando DHH y detectada por FAP, equilibra la adipogénesis a través de TIMP3 y la miogénesis a través de GDF10 (Fig. 7e).
- Distribución de relaciones públicas y contenido potenciado por SEO. Consiga amplificado hoy.
- PlatoData.Network Vertical Generativo Ai. Empodérate. Accede Aquí.
- PlatoAiStream. Inteligencia Web3. Conocimiento amplificado. Accede Aquí.
- PlatoESG. Automoción / vehículos eléctricos, Carbón, tecnología limpia, Energía, Ambiente, Solar, Gestión de residuos. Accede Aquí.
- Desplazamientos de bloque. Modernización de la propiedad de compensaciones ambientales. Accede Aquí.
- Fuente: https://www.nature.com/articles/s41467-023-39506-1

