Estrategia de diseño computacional.
Se diseñaron bucles cortos para conectar hélices de pRO-2.3 en una sola cadena utilizando una base de datos exhaustiva de muestras de columna vertebral compuestas de fragmentos que abarcan dos regiones helicoidales identificadas por DSSP en estructuras cristalográficas de alta resolución (como se describió anteriormente).14). Los bucles se identificaron en esta base de datos mediante una alineación rígida de los residuos terminales del fragmento y el objetivo utilizando un algoritmo de superposición optimizado.15. Los candidatos que cumplieron con una tolerancia de alineación de 0.35 Å RMSD se alinearon con la columna vertebral objetivo mediante coordenadas espaciales de torsión y restricciones de coordenadas suaves con las coordenadas de átomos pesados de la columna vertebral candidata alineada. Luego, las secuencias de bucle candidatas se diseñaron bajo restricciones de perfil de secuencia generadas mediante la alineación de la columna vertebral del bucle con la base de datos de la estructura de origen. Los candidatos con las puntuaciones más bajas fueron seleccionados para el diseño del circuito final.
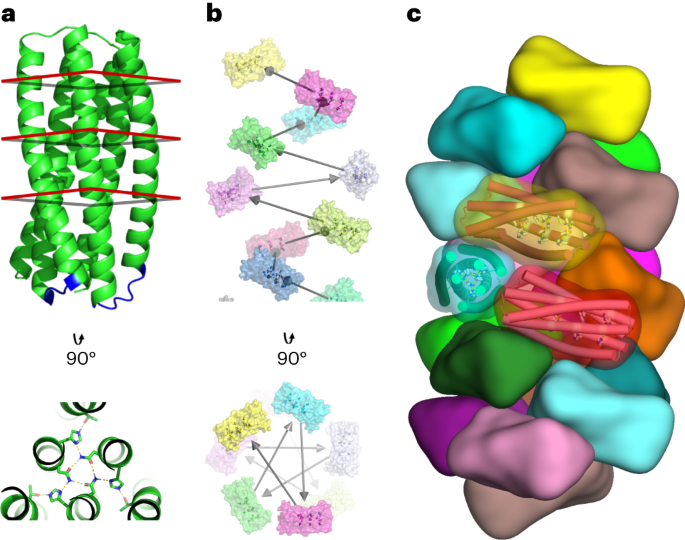
Métodos de diseño y acoplamiento helicoidal.7 Se aplicaron al pRO-2.3 vinculado para generar modelos de diseño de filamentos helicoidales. Los siguientes criterios filtraron las trayectorias de diseño individuales: una discrepancia superior a −15.0 unidades de energía Rosetta entre los estados ligado (polimérico) y no ligado (monomérico), un área de superficie de interfaz superior a 700 Å2, una complementariedad de la forma de Rosetta superior a 0.62 y un recuento de residuos polares insatisfechos inferior a 5. Los diseños que cumplían estos criterios se sometieron a un refinamiento manual, que implicaba reversiones de un solo punto a mutaciones consideradas no contribuyentes a la estabilización del estado ligado de la interfaz. Luego, el diseño con la puntuación más alta para cada configuración acoplada se integró en un conjunto de proteínas finalizado para su validación experimental.
Expresión y purificación de proteínas.
Los genes sintéticos de un total de 18 diseños se optimizaron para su expresión en Escherichia coli y adquirido de IDT, luego insertado en el sitio de clonación múltiple del vector pET29b+ entre los sitios de restricción NdeI y XhoI. Estas construcciones se introdujeron en BL21* (DE3) E. coli células competentes. Los transformantes se cultivaron en 50 ml de medio Terrific Broth suplementado con 200 mg l-1 kanamicina. La expresión, bajo el control de un promotor T7, se desarrolló durante 24 h a 37 °C utilizando la autoinducción de Studier.16 hasta que los cultivos fueron cosechados por centrifugación. Los sedimentos celulares se resuspendieron en solución salina tamponada con Tris (TBS) y se lisaron con detergente Bugbuster. La fracción soluble, clarificada por centrifugación, se purificó mediante Ni.2+ Cromatografía de afinidad de metales inmovilizados utilizando resina Ni-NTA Superflow. La resina con el lisado celular unido se lavó con diez volúmenes de columna de imidazol 40 mM y NaCl 500 mM, seguido de elución con imidazol 400 mM y NaCl 75 mM. Las fracciones solubles e insolubles se sometieron a análisis de electroforesis en gel de poliacrilamida-SDS. Se eligieron muestras que presentaban bandas de proteínas con el peso molecular correcto para el examen por microscopía electrónica. Los diseños seleccionados se ampliaron hasta 0.5 l para una mayor caracterización, y la expresión se realizó nuevamente durante 24 h a 37 °C utilizando la autoinducción de Studier.16 antes de la cosecha por centrifugación. Los sedimentos celulares se resuspendieron en TBS y se lisaron mediante microfluidización, seguido de purificación como se describe anteriormente.
Tinción negativa EM
Las fracciones solubles se concentraron en TBS (tampón Tris 25 mM, NaCl 75 mM, pH 8) para el cribado por microscopía electrónica. Se aplicó una gota de 6 µl (muestra de 1 µl diluida instantáneamente con 5 µl de tampón) sobre rejillas de cobre de malla 200 recubiertas de carbón con descarga luminosa negativa, se lavó con agua Milli-Q y se tiñó usando formiato de uranilo al 0.75% (pH 4.0). ) o Nano-W (pH 6.8) adquirido de Nanoprobes, Inc. como se describió anteriormente17. El cribado se realizó utilizando un microscopio electrónico de transmisión (FEI) Morgagni M100 de 268 kV o un microscopio electrónico de transmisión Talos L120C de 120 kV (ThermoFisher). Las imágenes se capturaron utilizando un sistema de cámara Teitz CMOS 4k de montaje inferior y se procesaron para mejorar el contraste utilizando el software Fiji (versión: 2.14.0/1.54f).18 para mayor claridad.
Las longitudes de las fibras se cuantificaron utilizando el algoritmo de rastreo de fibras en cryoSPARC.8. Este método identifica fibras mediante correlación cruzada con una clase de plantilla y rastreando fibras contiguas a partir de las partículas identificadas. Se utilizó una clase de plantilla generada a partir de DpHF19 para todas las fibras medidas. Las fibras se filtraron según la curvatura promedio (<0.0005 Å-1) y la correlación cruzada normalizada promedio (>0.5) en cada fibra. Para DpHF18, utilizamos 5, 2, 3, 20, 28 y 21 imágenes para pH 3, 3.5, 4.2, 5, 8 y 3 a 8, respectivamente. Para DpHF19, utilizamos 7, 8, 8, 28, 4 y 5 imágenes para pH 3, 3.5, 4.2, 5, 8 y 3 a 8, respectivamente. Para DpHF19_9his, utilizamos 6, 6, 8, 14, 15, 8 y 4 imágenes para pH 3, 3.5, 4.2, 5, 6, 8 y 3 a 8, respectivamente.
Crio-EM
Las muestras crio-EM se prepararon aplicando proteína a rejillas de carbono con agujeros CFLAT, secando el líquido y sumergiendo las rejillas en etano líquido usando un Vitrobot (ThermoFisher). Para DpHF19, los videos se adquirieron en un microscopio Glacios (ThermoFisher) equipado con una cámara K-2 Summit Direct Detect (Gatan Inc.) operando en modo de conteo, con un tamaño de píxel de 1.16 Å por píxel, 50 fotogramas y una dosis total de electrones. de 65 Å-2. Para DpHF18 y DpHF7, los videos se adquirieron en un Titan Krios (ThermoFisher) equipado con una cámara K-2 Summit Direct Detect (Gatan Inc.) operando en modo de superresolución, con un tamaño de píxel de 0.525 Å por píxel, 50 fotogramas y una dosis total de electrones de 90 Å-2. La recopilación de datos automatizada se realizó utilizando Leginon.19 versión 3.4. El procesamiento de datos se realizó utilizando cryoSPARC.8y los flujos de trabajo se resumen en las figuras complementarias. 10–12. Los videos se alinearon mediante corrección de movimiento de parches, con videos de súper resolución agrupados en un tamaño de píxel de 1.05 Å. Los parámetros de la función de transferencia de contraste (CTF) se estimaron utilizando el parche CTF. Se realizó un rastreo de filamentos sin plantilla en un subconjunto de imágenes y las partículas resultantes se sometieron a una clasificación 2D. Luego se utilizaron clases 2D seleccionadas como plantillas para el rastreo de filamentos basado en plantillas en conjuntos de datos completos. Después de múltiples rondas de clasificación 2D, las partículas seleccionadas se sometieron a un refinamiento 3D con simetría helicoidal impuesta y un refinamiento no uniforme habilitado. Para DpHF19, impusimos simetría helicoidal de un inicio que relaciona subunidades individuales sin contacto, en lugar de los parámetros de simetría helicoidal de dos inicios. Para DpHF7 y DpHF19, también se refinaron el desenfoque por partícula, la inclinación del haz y la aberración esférica. La modificación de la densidad se realizó utilizando ResolveCryoEM en Phenix.20,21 versión phenix-1.20.1. Los modelos atómicos para DpHF18 y DpHF19 se refinaron en mapas crio-EM utilizando ISOLDE22, seguido del refinamiento del espacio real en Phenix, con las restricciones de rotámero y Ramachandran deshabilitadas y con restricciones de referencia impuestas por el modelo inicial de entrada. La elucidación del modelo para DpHF7 empleó el protocolo de construcción de modelos de novo en la densidad de unidades asimétricas crio-EM segmentadas.23. La incorporación y el refinamiento de residuos posteriores se lograron utilizando RosettaCM.24 versión 2019.31, que aprovecha la simetría en el mapa crio-EM no segmentado para un ajuste óptimo a la densidad y interfaces intrafilamento. Se realizó una ronda final de refinamiento del espacio real en Phenix, como se describió anteriormente para DpHF18 y DpHF19. Las estadísticas de recopilación, refinamiento y validación de datos de Cryo-EM se resumen en la tabla complementaria 1.
TIRFM
Conjunto de fibras
Para obtener imágenes de la nucleación sembrada de fibras sensibles al pH, se marcaron fibras DpHF18 con dos fluoróforos diferentes conjugados con maleimida, Oregon488 y sulfo-Cy5. Las fibras se marcaron con un exceso molar de 10x, en PBS + TCEP 1 mM durante 4 h a temperatura ambiente, antes del intercambio de tampón en TBS (Tris 25 mM, NaCl 100 mM, pH 8.0) en una columna de centrifugación Zeba y concentración a 30 μM. . Las fibras verdes a 30 µM se desmontaron mediante la adición de citrato 1 M (0.6 µl de citrato por 20 µl de fibras) para reducir el pH a 3.0. La solución se incubó durante 5 minutos antes de la adición de Tris (3.6 µl de solución madre 1 M) para devolver el pH a 8.0; Se añadió a la solución 1 μl de fibras DpHF18-Cy5 ensambladas a 30 μM. Posteriormente, la solución se incubó a temperatura ambiente antes de la centrifugación a 13,000 g durante 2 min en una centrífuga de mesa. Las fibras se resuspendieron en TBS y se tomaron imágenes mediante TIRFM.
Desmontaje de fibra
Se realizaron imágenes TIRFM rápidas de fibras desmontadas a pH bajo en un sistema TIRF personalizado basado en un soporte Nikon Ti equipado con un sistema de enfoque perfecto junto con una rápida Z etapa piezoeléctrica (ASI), un iluminador TIRF azimutal (iLas2, Roper France) con un campo de visión extendido personalizado (Cairn) y un objetivo PLAN Apo 1.45 NA ×100. Las imágenes se adquirieron con una cámara sCMOS retroiluminada Photometrics Prime 95B ejecutada en modo de obturador pseudo global, sincronizada con la iluminación azimutal. El sistema fue operado por Metamorph 7.10.1.161. Se tomaron imágenes de fibras marcadas con maleimida con sulfo-Cy5 con un láser de 630 nm (OBIS coherente de 150 mW montado en un lanzamiento láser Cairn) y se tomaron imágenes usando un filtro Chroma ET655lp montado en una rueda Cairn Optospin a una velocidad de fotogramas de 1 fotograma cada 16 ms.
Se tomaron imágenes de las fibras en un tampón de imágenes (Tris 25 mM, pH 8.0, NaCl 100 mM) en una celda de flujo Ibidi montada sobre cubreobjetos aptos para sala limpia (personalizados, 25 × 75 mm).2, Nexterion) y pasivado con PLL-PEG (0.1 mg ml-1 en Hepes 20 mM, pH 7.6; 5 minutos). Se permitió que las fibras se depositaran en el cubreobjetos durante 5 minutos antes de eliminar las fibras sueltas con el tampón de imágenes. Durante la adquisición rápida, el pH se redujo haciendo fluir en un tampón de pH bajo (Tris 25 mM, NaCl 100 mM, pH 3.0).
Para medir el desmontaje de las fibras en solución a granel, las fibras preformadas en tubos Eppendorf de 1.5 ml se intercambiaron por tampones de citrato a un pH más bajo para estimular el desmontaje. Se eliminó una porción de cada reacción de pH en varios puntos de tiempo y se añadió a una placa de 96 pocillos y durante 10 minutos para permitir que las fibras se sedimentaran y se adhirieran al sustrato de vidrio. Para cada condición y punto temporal, se adquirieron nueve campos de visión en un microscopio IN Cell Analyzer 2500HS (Molecular Devices) utilizando un objetivo de aire Nikon ×60 PLAN Apo 0.95 NA y una fuente de excitación LED de 631 nm, tiempo de exposición de 150 ms con emisión recopilada. a través de un filtro de paso de banda de 684 ± 24 nm. Las imágenes se cuantificaron utilizando un script CellProfiler personalizado para segmentar fibras con el algoritmo de umbral de Otsu.25. Los límites superior e inferior del umbral, así como la ventana adaptativa para la identificación del objeto, se ajustaron hasta que las fibras se identificaron correctamente en relación con la señal de fondo. La longitud del eje principal de los objetos identificados mediante la tubería CellProfiler se representó frente al tiempo de incubación para cada condición de pH.
AFM en fase líquida
preparación de la muestra
Incubamos 10 µl de una solución de polilisina al 0.01% en peso sobre una superficie de mica moscovita recién escindida (12 mm, Ted Pella Inc.) durante 2 minutos. Se eliminó el exceso de solución y la superficie se enjuagó con agua y se secó con N2 Natural7. Luego se incubaron 30 µl de solución de proteína 10 µM en el tampón de imágenes (Tris-HCl 25 mM, NaCl 400 mM a pH 8) sobre la mica recubierta de polilisina durante 30 minutos y se lavaron con el tampón de imágenes para eliminar el exceso de proteína. El pH del tampón de desmontaje (Tris-HCl 25 mM, NaCl 400 mM, pH 4.1, 4.4, 4, 5 o 4.7) se ajustó con NaOH 10 M o ácido cítrico 1 M y se filtró con un filtro de PVDF con un tamaño de poro de 0.1 µm antes de su uso. . Para experimentos con fotoácidos, se incubó una solución de proteína 10 µM en Tris-HCl 25 mM, pH 8, sobre mica desnuda durante 30 minutos y se lavó con Tris-HCl 25 mM, pH 5.5; Se llevó a cabo una etapa adicional de deposición y enjuague si la densidad numérica de fibras en la superficie era baja. También preparamos recientemente 1-nitrobenzaldehído 2 mM (Sigma-Aldrich) en Tris-HCl 25 mM, pH 5.5 y lo usamos inmediatamente sin exposición a la luz en ningún momento.26. Las mediciones espectroscópicas y de pH indicaron que el 2-nitrobenzaldehído se puede activar entre longitudes de onda de 200 y 405 nm y reduce el pH de 5.5 a 2.7, y que una mayor intensidad del láser conduce a un consumo y acidificación más rápidos.
Proyección de imagen
Para el estudio cinético a composición constante, los sustratos de polilisina mica recubiertos de proteínas se colocaron debajo de la celda líquida AFM (Bruker Multimode8). Las imágenes se capturaron en el búfer de imágenes utilizando un voladizo de nitruro de silicio limpio (Bruker, SNL-10, constante de resorte: 0.12 N m-1, UV ozonizado durante 5 min) en modo de golpeteo a temperatura ambiente (25 °C). Antes de hacer fluir el búfer de desmontaje, se tomaron imágenes de las fibras de forma continua durante 10 minutos para optimizar los parámetros (256 líneas de exploración, velocidad de exploración de 1.5 Hz, ganancia integral alta (3–4) y amplitud libre de 50–100 mV). Después de confirmar que no se produjo ningún daño inducido por el voladizo, se inyectó continuamente el tampón de desmontaje a 25 µl min.-1. La configuración de flujo se optimizó para proporcionar un tiempo de residencia insignificante y un cambio rápido de pH.10.
Para el estudio del fotoácido, se colocó mica recubierta de proteína con Tris-HCl 25 mM, pH 5.5, debajo de la celda líquida de un Cypher VRS AFM (Asylum Research) equipado con láser BlueDrive (filtro de intensidad ×0.3, longitud de onda de 405 nm) con válvula de ventilación. abierto y operado en modo de golpeteo. Después de confirmar la alta cobertura de la superficie de las fibras, el tampón de imágenes se reemplazó con 1-nitrobenzaldehído 2 mM en Tris-HCl 25 mM, pH 5.5, se operó sin exposición a luz de fondo visible y se tomaron imágenes nuevamente. Luego se retrajo el voladizo, se encendió BlueDrive y se rastreó repetidamente en áreas preseleccionadas utilizando el microscopio óptico motorizado del AFM. El tiempo total de exposición a los rayos UV durante la trama/permanencia para patrones de puntos y líneas no fue más de 10 minutos, después de lo cual el voladizo se movió nuevamente a las áreas expuestas y se tomaron imágenes. Para los cambios globales de pH, la ventana de cuarzo de la celda líquida AFM en contacto con la solución de fotoácido se expuso a una lámpara UV de mano (longitud de onda de 364 nm) durante 7 minutos y luego se tomaron imágenes.
Las imágenes se procesaron con el software de análisis de datos Gwyddion SPM v2.62 y se analizaron con el software Fiji v1.53s.18. Para la cinética, se midió la longitud total de la fibra y se excluyeron de la medición de la longitud los fragmentos considerados ya desensamblados. Para medir la tasa de desmontaje en cada extremo de las fibras individuales (Figura complementaria. 8), el centro de la fibra (la mitad de la longitud inicial) se asignó como segundo extremo para medir la longitud, mientras que para los fragmentos de fibra, el centro del fragmento se midió como segundo extremo.
- Distribución de relaciones públicas y contenido potenciado por SEO. Consiga amplificado hoy.
- PlatoData.Network Vertical Generativo Ai. Empodérate. Accede Aquí.
- PlatoAiStream. Inteligencia Web3. Conocimiento amplificado. Accede Aquí.
- PlatoESG. Carbón, tecnología limpia, Energía, Ambiente, Solar, Gestión de residuos. Accede Aquí.
- PlatoSalud. Inteligencia en Biotecnología y Ensayos Clínicos. Accede Aquí.
- Fuente: https://www.nature.com/articles/s41565-024-01641-1

