Las proteínas impulsan muchos procesos biológicos, como la actividad enzimática, el transporte molecular y el soporte celular. La estructura tridimensional de una proteína proporciona información sobre su función y cómo interactúa con otras biomoléculas. Los métodos experimentales para determinar la estructura de las proteínas, como la cristalografía de rayos X y la espectroscopia de RMN, son costosos y consumen mucho tiempo.
Por el contrario, los métodos computacionales desarrollados recientemente pueden predecir con rapidez y precisión la estructura de una proteína a partir de su secuencia de aminoácidos. Estos métodos son fundamentales para las proteínas que son difíciles de estudiar experimentalmente, como las proteínas de membrana, los objetivos de muchos fármacos. Un ejemplo bien conocido de esto es AlphaFold, un algoritmo basado en el aprendizaje profundo celebrado por sus predicciones precisas.
ESMFold es otro método altamente preciso basado en el aprendizaje profundo desarrollado para predecir la estructura de la proteína a partir de su secuencia de aminoácidos. ESMFold utiliza un modelo de lenguaje de proteínas grandes (pLM) como columna vertebral y opera de extremo a extremo. A diferencia de AlphaFold2, no necesita una búsqueda o Alineación de secuencias múltiples (MSA), ni depende de bases de datos externas para generar predicciones. En cambio, el equipo de desarrollo entrenó el modelo en millones de secuencias de proteínas de UniRef. Durante el entrenamiento, el modelo desarrolló patrones de atención que representan elegantemente las interacciones evolutivas entre los aminoácidos en la secuencia. Este uso de un pLM en lugar de un MSA permite tiempos de predicción hasta 60 veces más rápidos que otros modelos de última generación.
En esta publicación, usamos el modelo ESMFold preentrenado de Hugging Face con Amazon SageMaker para predecir la estructura de la cadena pesada de trastuzumab, anticuerpo monoclonal desarrollado por primera vez por Genentech para el tratamiento de Cáncer de mama positivo para HER2. La predicción rápida de la estructura de esta proteína podría ser útil si los investigadores quisieran probar el efecto de las modificaciones de secuencia. Esto podría potencialmente conducir a una mejor supervivencia del paciente o menos efectos secundarios.
Esta publicación proporciona un ejemplo de cuaderno Jupyter y scripts relacionados a continuación Repositorio GitHub.
Requisitos previos
Recomendamos ejecutar este ejemplo en un Estudio Amazon SageMaker cuaderno ejecutando la imagen optimizada para CPU PyTorch 1.13 Python 3.9 en un tipo de instancia ml.r5.xlarge.
Visualice la estructura experimental de trastuzumab
Para empezar, usamos el biopython librería y una secuencia de comandos de ayuda para descargar la estructura de trastuzumab del Banco de datos de proteínas RCSB:
A continuación, usamos el py3Dmol biblioteca para visualizar la estructura como una visualización 3D interactiva:
La siguiente figura representa la estructura de proteína 3D 1N8Z del Protein Data Bank (PDB). En esta imagen, la cadena ligera de trastuzumab se muestra en naranja, la cadena pesada en azul (con la región variable en azul claro) y el antígeno HER2 en verde.

Primero usaremos ESMFold para predecir la estructura de la cadena pesada (Cadena B) a partir de su secuencia de aminoácidos. Luego, compararemos la predicción con la estructura determinada experimentalmente que se muestra arriba.
Prediga la estructura de la cadena pesada de trastuzumab a partir de su secuencia usando ESMFold
Usemos el modelo ESMFold para predecir la estructura de la cadena pesada y compararla con el resultado experimental. Para comenzar, usaremos un entorno de cuaderno preconstruido en Studio que viene con varias bibliotecas importantes, como PyTorch, pre instalado. Aunque podríamos usar un tipo de instancia acelerada para mejorar el rendimiento de nuestro análisis de notebook, en su lugar usaremos una instancia no acelerada y ejecutaremos la predicción ESMFold en una CPU.
En primer lugar, cargamos el tokenizador y el modelo ESMFold preentrenado desde abrazando la cara hub:
A continuación, copiamos el modelo a nuestro dispositivo (CPU en este caso) y configuramos algunos parámetros del modelo:
Para preparar la secuencia de proteínas para el análisis, necesitamos tokenizarla. Esto traduce los símbolos de aminoácidos (EVQLV...) a un formato numérico que el modelo ESMFold puede entender (6,19,5,10,19,...):
A continuación, copiamos la entrada tokenizada al modo, hacemos una predicción y guardamos el resultado en un archivo:
Esto toma alrededor de 3 minutos en un tipo de instancia no acelerada, como un r5.
Podemos verificar la precisión de la predicción ESMFold comparándola con la estructura experimental. Esto lo hacemos usando el Alinear EE. UU. herramienta desarrollada por Zhang Lab en la Universidad de Michigan:
| PDBcadena1 | PDBcadena2 | Puntuación TM |
| datos/predicción.pdb:A | datos/experimental.pdb:B | 0.802 |
La puntaje de modelado de plantilla (TM-score) es una métrica para evaluar la similitud de las estructuras de proteínas. Una puntuación de 1.0 indica una coincidencia perfecta. Las puntuaciones superiores a 0.7 indican que las proteínas comparten la misma estructura principal. Las puntuaciones superiores a 0.9 indican que las proteínas son funcionalmente intercambiable para uso aguas abajo. En nuestro caso de lograr TM-Score 0.802, la predicción ESMFold probablemente sería apropiada para aplicaciones como la puntuación de estructuras o los experimentos de unión de ligandos, pero puede no ser adecuada para casos de uso como reemplazo molecular que requieren una precisión extremadamente alta.
Podemos validar este resultado visualizando las estructuras alineadas. Las dos estructuras muestran un alto, pero no perfecto, grado de superposición. ¡Las predicciones de la estructura de proteínas son un campo en rápida evolución y muchos equipos de investigación están desarrollando algoritmos cada vez más precisos!
Implemente ESMFold como punto final de inferencia de SageMaker
Ejecutar la inferencia de modelos en un cuaderno está bien para la experimentación, pero ¿qué sucede si necesita integrar su modelo con una aplicación? ¿O una canalización de MLOps? En este caso, una mejor opción es implementar su modelo como punto final de inferencia. En el siguiente ejemplo, implementaremos ESMFold como punto final de inferencia en tiempo real de SageMaker en una instancia acelerada. Los terminales en tiempo real de SageMaker proporcionan una forma escalable, rentable y segura de implementar y alojar modelos de aprendizaje automático (ML). Con el escalado automático, puede ajustar la cantidad de instancias que ejecutan el punto final para satisfacer las demandas de su aplicación, optimizando los costos y asegurando una alta disponibilidad.
El pre-construido Contenedor de SageMaker para Hugging Face facilita la implementación de modelos de aprendizaje profundo para tareas comunes. Sin embargo, para casos de uso novedosos como la predicción de la estructura de proteínas, necesitamos definir un inference.py secuencia de comandos para cargar el modelo, ejecutar la predicción y formatear la salida. Este script incluye gran parte del mismo código que usamos en nuestro cuaderno. También creamos un requirements.txt para definir algunas dependencias de Python para que las use nuestro punto final. Puede ver los archivos que creamos en el Repositorio GitHub.
En la siguiente figura, las estructuras experimental (azul) y predicha (roja) de la cadena pesada de trastuzumab son muy similares, pero no idénticas.

Después de haber creado los archivos necesarios en el code directorio, implementamos nuestro modelo usando SageMaker HuggingFaceModel clase. Esto utiliza un contenedor prediseñado para simplificar el proceso de implementación de modelos Hugging Face en SageMaker. Tenga en cuenta que puede llevar 10 minutos o más crear el extremo, dependiendo de la disponibilidad de ml.g4dn tipos de instancias en nuestra región.
Cuando se completa la implementación del punto final, podemos volver a enviar la secuencia de la proteína y mostrar las primeras filas de la predicción:
Debido a que implementamos nuestro punto final en una instancia acelerada, la predicción solo debería tomar unos segundos. Cada fila en el resultado corresponde a un solo átomo e incluye la identidad del aminoácido, tres coordenadas espaciales y un puntuación pLDDT que representa la confianza de la predicción en esa ubicación.
| PDB_GROUP | ID | ÁTOMO_ETIQUETA | RES_ID | CADENA_ID | SEQ_ID | CARTON_X | CARTON_Y | CARTN_Z | OCUPACIÓN | PLDDT | ÁTOMO_ID |
| ATOM | 1 | N | GLU | A | 1 | 14.578 | -19.953 | 1.47 | 1 | 0.83 | N |
| ATOM | 2 | CA | GLU | A | 1 | 13.166 | -19.595 | 1.577 | 1 | 0.84 | C |
| ATOM | 3 | CA | GLU | A | 1 | 12.737 | -18.693 | 0.423 | 1 | 0.86 | C |
| ATOM | 4 | CB | GLU | A | 1 | 12.886 | -18.906 | 2.915 | 1 | 0.8 | C |
| ATOM | 5 | O | GLU | A | 1 | 13.417 | -17.715 | 0.106 | 1 | 0.83 | O |
| ATOM | 6 | cg | GLU | A | 1 | 11.407 | -18.694 | 3.2 | 1 | 0.71 | C |
| ATOM | 7 | cd | GLU | A | 1 | 11.141 | -18.042 | 4.548 | 1 | 0.68 | C |
| ATOM | 8 | OE1 | GLU | A | 1 | 12.108 | -17.805 | 5.307 | 1 | 0.68 | O |
| ATOM | 9 | OE2 | GLU | A | 1 | 9.958 | -17.767 | 4.847 | 1 | 0.61 | O |
| ATOM | 10 | N | VAL | A | 2 | 11.678 | -19.063 | -0.258 | 1 | 0.87 | N |
| ATOM | 11 | CA | VAL | A | 2 | 11.207 | -18.309 | -1.415 | 1 | 0.87 | C |
Usando el mismo método que antes, vemos que las predicciones del cuaderno y del punto final son idénticas.
| PDBcadena1 | PDBcadena2 | Puntuación TM |
| datos/endpoint_prediction.pdb:A | datos/predicción.pdb:A | 1.0 |
Como se observa en la siguiente figura, las predicciones de ESMFold generadas en el portátil (rojo) y por el punto final (azul) muestran una alineación perfecta.
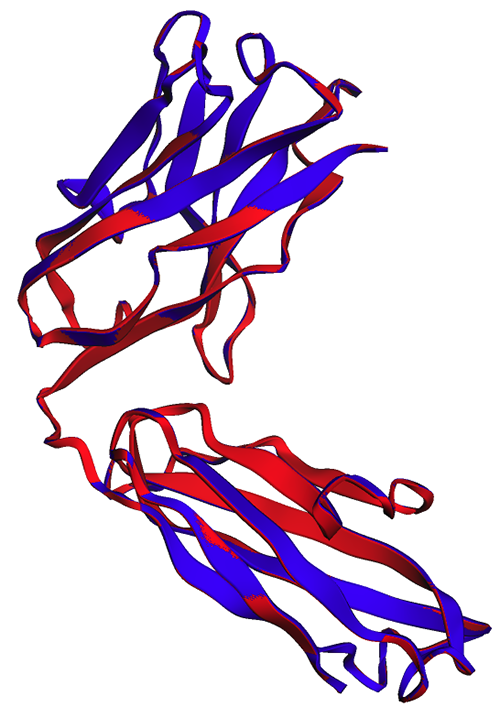
Limpiar
Para evitar cargos adicionales, eliminamos nuestro punto final de inferencia y los datos de prueba:
Resumen
La predicción computacional de la estructura de proteínas es una herramienta fundamental para comprender la función de las proteínas. Además de la investigación básica, los algoritmos como AlphaFold y ESMFold tienen muchas aplicaciones en medicina y biotecnología. Los conocimientos estructurales generados por estos modelos nos ayudan a comprender mejor cómo interactúan las biomoléculas. Esto puede conducir a mejores herramientas de diagnóstico y terapias para los pacientes.
En esta publicación, mostramos cómo implementar el modelo de lenguaje de proteínas ESMFold de Hugging Face Hub como punto final de inferencia escalable mediante SageMaker. Para obtener más información sobre la implementación de modelos Hugging Face en SageMaker, consulte Use Hugging Face con Amazon SageMaker. También puede encontrar más ejemplos de ciencia de proteínas en el Impresionante análisis de proteínas en AWS repositorio de GitHub. ¡Déjanos un comentario si hay otros ejemplos que te gustaría ver!
Acerca de los autores
 Brian leal es arquitecto sénior de soluciones de inteligencia artificial y aprendizaje automático en el equipo global de salud y ciencias biológicas de Amazon Web Services. Tiene más de 17 años de experiencia en biotecnología y aprendizaje automático, y le apasiona ayudar a los clientes a resolver desafíos genómicos y proteómicos. En su tiempo libre, le gusta cocinar y comer con sus amigos y familiares.
Brian leal es arquitecto sénior de soluciones de inteligencia artificial y aprendizaje automático en el equipo global de salud y ciencias biológicas de Amazon Web Services. Tiene más de 17 años de experiencia en biotecnología y aprendizaje automático, y le apasiona ayudar a los clientes a resolver desafíos genómicos y proteómicos. En su tiempo libre, le gusta cocinar y comer con sus amigos y familiares.
 Shamika Ariyawansa es Arquitecto de Soluciones Especializado en AI/ML en el equipo Global de Salud y Ciencias de la Vida en Amazon Web Services. Trabaja apasionadamente con los clientes para acelerar su adopción de IA y ML al brindar orientación técnica y ayudarlos a innovar y crear soluciones de nube seguras en AWS. Fuera del trabajo, le encanta esquiar y hacer todoterreno.
Shamika Ariyawansa es Arquitecto de Soluciones Especializado en AI/ML en el equipo Global de Salud y Ciencias de la Vida en Amazon Web Services. Trabaja apasionadamente con los clientes para acelerar su adopción de IA y ML al brindar orientación técnica y ayudarlos a innovar y crear soluciones de nube seguras en AWS. Fuera del trabajo, le encanta esquiar y hacer todoterreno.
 yanjun qi es gerente sénior de ciencias aplicadas en el laboratorio de soluciones de aprendizaje automático de AWS. Ella innova y aplica el aprendizaje automático para ayudar a los clientes de AWS a acelerar su adopción de la inteligencia artificial y la nube.
yanjun qi es gerente sénior de ciencias aplicadas en el laboratorio de soluciones de aprendizaje automático de AWS. Ella innova y aplica el aprendizaje automático para ayudar a los clientes de AWS a acelerar su adopción de la inteligencia artificial y la nube.
- Distribución de relaciones públicas y contenido potenciado por SEO. Consiga amplificado hoy.
- PlatoAiStream. Inteligencia de datos Web3. Conocimiento amplificado. Accede Aquí.
- Acuñando el futuro con Adryenn Ashley. Accede Aquí.
- Compra y Vende Acciones en Empresas PRE-IPO con PREIPO®. Accede Aquí.
- Fuente: https://aws.amazon.com/blogs/machine-learning/accelerate-protein-structure-prediction-with-the-esmfold-language-model-on-amazon-sagemaker/

