SNIP1 es necesario para la supervivencia de NPC en el cerebro embrionario murino
Primero examinamos la expresión de recorte1 en el cerebro embrionario murino por RNAscope40. En el día embrionario E11.5 y E13.5, recorte1 Las transcripciones se expresaron en casi todas las células y se expresaron de manera sólida en el neuroepitelio que recubre los ventrículos, donde residen los NPC (Fig. 1b, c). Para estudiar SNIP1 en el cerebro embrionario murino, utilizamos Nestín (Nes) ::Cre para agotar condicionalmente SNIP1 en NPC, en lo sucesivo denominado recorte1Nes–KO. Nes::Cre se expresa en NPC para recombinar los sitios flox y eliminar el exón 2 de recorte1 (Higo. 1a, Fig. Suplementaria 1d-f, Fig. Suplementaria 2a–d). por E15, recorte1Nes-Los embriones KO mostraron adelgazamiento severo de los tejidos cerebrales y displasia con una penetrancia del 100% (Fig. 1b, c, p < 0.0001 por la prueba exacta de Fisher). Comprender los fundamentos celulares de la displasia cerebral en recorte1Nes–KO, examinamos la proliferación celular y la apoptosis de los NPC. Los NPC se identificaron por su expresión del marcador de células madre neurales SRY-box 2 (SOX2). Para identificar las células en proliferación in vivo, inyectamos BrdU en madres preñadas y/o detectamos el marcador proliferativo Ki67. La cuantificación de estos marcadores en el neuroepitelio no reveló una diferencia significativa en los NPC proliferativos en el control de hermanos y recorte1Nes–Embriones KO (Fig. 2e-h).
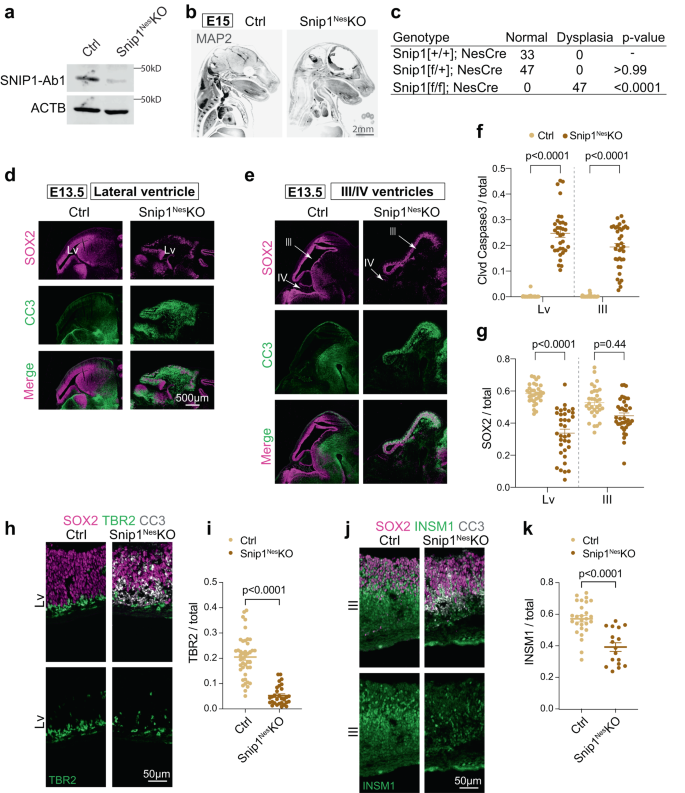
a WB de control y recorte1Nes-KO NPC en E13.5. Al menos 5 repeticiones de control y recorte1Nes-Los PNJ KO mostraron resultados similares. SNIP1-Ab1; anticuerpo anti-SNIP1 de ProteinTech. b SI de MAP2 en control y recorte1Nes-Embriones KO en E15. Los embriones se aclararon mediante el método iDISCO y se tomaron imágenes con microscopía de hoja de luz. Dos réplicas de IF mostraron resultados similares. Barra, 2 mm. c Penetración de displasia cerebral en embriones E13.5. La displasia cerebral se determinó por el adelgazamiento del tejido cerebral. La significación estadística se calculó mediante la prueba exacta de Fisher. Delaware IF de SOX2 y caspasa 3 escindida (CC3) en criosecciones sagitales del cerebro E13.5. Se examinaron las zonas germinales alrededor del ventrículo lateral (Lv, cerebro anterior), tercer ventrículo (cerebro medio) y cuarto (cerebro posterior). Barra, 500 μm. f, g Cuantificación de células CC3 positivas y SOX2 positivas en el revestimiento neuroepitelial de los ventrículos de control y recorte1Nes-Embriones KO en E13.5. Se usó tinción DAPI para contar el número total de células. Cada punto de datos representa una imagen. Ocho embriones de control y 7 recorte1Nes-Se analizaron embriones KO. En (f), para ventrículo lateral, n = 38 imágenes (control) y n = 34 (recorte1Nes-KO); para el tercer ventrículo, n = 38 (control) y n = 36 (recorte1Nes-KO). En (g), para ventrículo lateral, n = 33 (control) y n = 35 (recorte1Nes-KO); para el tercer ventrículo, n = 32 (control) y n = 37 (recorte1Nes-KO). Los datos se presentan como media ± SEM, y se utilizó ANOVA de dos vías para el análisis estadístico. h – j IF de SOX2 y CC3 superpuesto con marcadores de linaje neuronal TBR2 e INSM1 del cerebro E13.5. Barra, 50 μm. i, k Cuantificación de células TBR2 positivas o INSM1 positivas en el revestimiento neuroepitelial de los ventrículos laterales o terceros. Cada punto de datos representa una imagen. Cinco a 8 embriones de control y 3-7 recorte1Nes-Se analizaron embriones KO. En (i), n = 43 imágenes (control) y n = 30 (recorte1Nes-KO); en (k), n = 27 (control) y n = 17 (recorte1Nes-KO). Los datos se presentan como media ± SEM, y se utilizó ANOVA de dos vías para el análisis estadístico. Los datos de origen se proporcionan en un archivo de datos de origen (a, f, g, i, k).
A continuación, probamos la apoptosis por IF de (cl) -caspasa 3 escindida. En E13.5, todos los ventrículos de recorte1Nes–KO mostró una fuerte inducción de cl-caspasa 3 en las zonas subventriculares del neuroepitelio (Fig. 1d-f). Las señales de cl-caspasa 3 se superpusieron a los NPC positivos para SOX2 y a los progenitores intermedios positivos para TBR2 (Fig. 2i–l), que se redujeron notablemente en recorte1Nes–Neuroepitelio KO (Fig. 1d-yo). Los progenitores intermedios positivos para INSM1 se redujeron de manera similar en recorte1Nes–cerebro medio KO (Fig. 1j,k). Estos datos sugieren que SNIP1 suprime la apoptosis en NPC y progenitores intermedios en el cerebro en desarrollo.
Para capturar los eventos anteriores de la recorte1Nes–Cerebro KO, examinamos cl-caspasa 3 en el cerebro embrionario E11.5. Para E11.5, se detectaron señales de cl-caspasa 3 en todo el recorte1Nes–Cerebro KO (Fig. 3a–d). Como Nes::Cre está encendido por E10.541, el agotamiento de SNIP1 probablemente induce la apoptosis dentro de las 24 h. La cuantificación de células positivas para SOX2 en los ventrículos lateral, tercero y cuarto mostró que solo las NPC en el cuarto ventrículo se agotaron por E11.5 (Fig. 3e). De aquí en adelante, enfocamos nuestros análisis de los ventrículos laterales y terceros para estudiar el control de la apoptosis por SNIP1.
Para determinar que el agotamiento de SNIP1 causa constantemente la apoptosis observada, utilizamos emx1:: Cre para agotar condicionalmente SNIP1 en NPC del telencéfalo dorsal42. recorte1emx1–Los embriones KO también mostraron una fuerte inducción de apoptosis, pérdida de progenitores intermedios positivos para TBR2 y displasia del cerebro anterior (Fig. 3f-k). Estos hallazgos respaldan que en el cerebro murino en desarrollo, la inducción de apoptosis y la displasia son específicas del agotamiento de SNIP1. Estos datos respaldan aún más nuestra conclusión de que se requiere SNIP1 para un mecanismo antiapoptótico y pro-supervivencia en NPC y progenitores intermedios.
SNIP1 suprime el programa de apoptosis intrínseco
Para arrojar luz sobre los fundamentos moleculares de la defectuosa recorte1Nes–KO NPC, realizamos secuenciación de ARN (RNA-seq) de NPC positivos para SOX2 clasificados de E13.5 recorte1Nes–KO y controles hermanos (Fig. 2a, Fig. Suplementaria 4a). Analizamos genes con valores de recuento por millón (CPM) > 1 en control o recorte1Nes–NPC KO. Usando el criterio de tasa de descubrimiento falso (FDR) <0.05 para comparar conjuntos de datos de 4 réplicas, cada uno de control y recorte1Nes-KO NPC, identificamos 1210 genes regulados al alza y 1621 genes regulados a la baja en recorte1Nes-KO (fig. 2b, Fig. Suplementaria 4b–d).
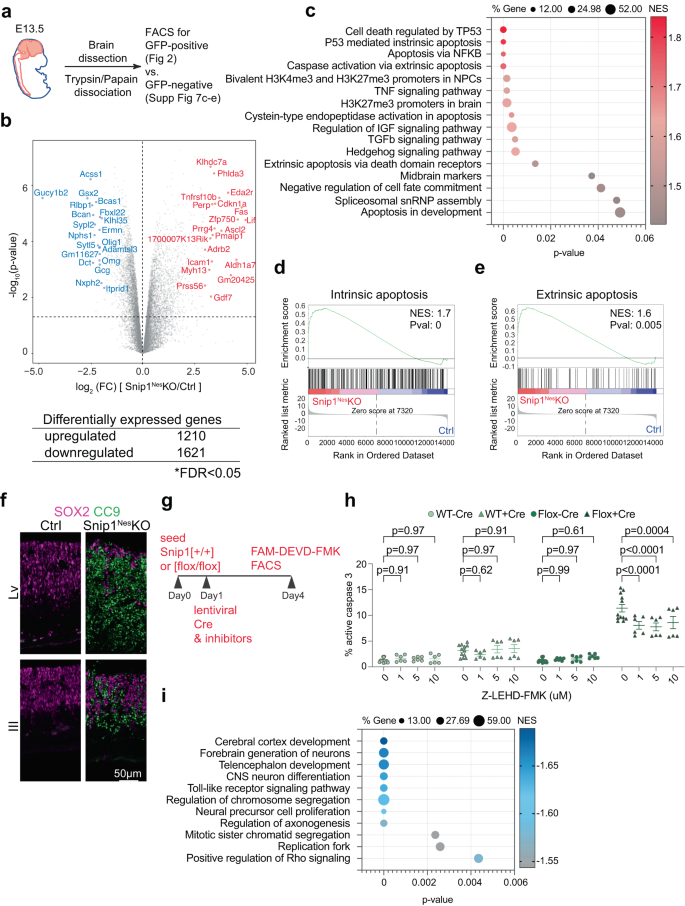
a Esquema de la colección de NPC cerebrales. b Gráfico de volcán y el número de genes expresados diferencialmente entre el control y recorte1Nes-KO NPC. La tabla muestra el número de genes que pasaron el límite de FDR < 0.05. FDR se calculó por el método de Benjamini & Hochberg. P los valores se calcularon mediante Voom-limma de dos caras t . c, i Gráficos de burbujas de los conjuntos de genes enriquecidos en genes regulados al alza y genes regulados a la baja en recorte1Nes-KO contra NPC de control. Los genes expresados diferencialmente se clasificaron primero por su cambio de pliegue, p-valor y nivel de expresión antes de realizar el análisis de enriquecimiento de conjuntos de genes (GSEA). P-los valores se calcularon mediante una prueba de permutación del lado derecho con ajuste FDR. d, e GSEA representativo de genes regulados al alza en recorte1Nes-KO contra NPC de control. Los genes regulados al alza se enriquecieron en conjuntos de genes relacionados con la apoptosis tanto intrínseca como extrínseca. Los genes expresados diferencialmente se clasificaron primero por su cambio de pliegue y p-valor antes de que se realizara GSEA. f IF de caspasa 9 escindida (CC9) superpuesta con SOX2 en criosecciones sagitales del cerebro E13.5. Barra, 50 μm. g Esquema de transducción con mCherry-Cre lentivirus y tratamiento con inhibidores en recorte1[+/+] y recorte1[flox/flox] PNJ. h El porcentaje de células con caspasa 3 activa cuantificado por FACS. El inhibidor de caspasa 9 (Z-LEHD-FMK) se agregó a diferentes concentraciones junto con el lentivirus mCherry-Cre. Se muestra el porcentaje de población positiva para FAM-FLICA (caspasa 3 activa) (sobre la población total). N = 12 para control DMSO y n = 6 para el resto de la muestra. Los datos se presentan como media ± SEM, y se utilizó ANOVA de dos vías para el análisis estadístico. La estrategia de activación y los gráficos FACS representativos se muestran en Información complementaria (Fig. 4e, higo suplementario 14a). Los datos de origen se proporcionan en un archivo de datos de origen (c, h, i).
El análisis de enriquecimiento de conjuntos de genes (GSEA) reveló que los genes regulados al alza en recorte1Nes–Los NPC KO se enriquecieron en funciones relacionadas con la apoptosis mediada por p53, H3K27me3 o promotores bivalentes en los NPC y el cerebro, marcadores del mesencéfalo, partículas pequeñas de ribonucleoproteína nuclear esplicosomal y vías de señalización que involucran TNF, IGF, TGFβ y Hedgehog (Fig. 2c). A medida que las firmas apoptóticas intrínsecas y extrínsecas se enriquecieron en los genes regulados al alza en recorte1Nes–PNJ KO (Fig. 2d, e), examinamos la activación de estas dos vías. IF mostró poca señal de cl-caspasa 8, un efector de la vía de apoptosis extrínseca43,44 (Fig. complementaria. 5a) pero fuertes señales de cl-caspasa 9, un efector de la vía de apoptosis intrínseca45,46,47, en todo el neuroepitelio de los ventrículos (Fig. 2f). Además, cuantificación de cl-caspasa 3 por FACS de tinción FAM-DEVD-FMK48,49 muestran que mientras que la inhibición de la caspasa 8 alteró modestamente la apoptosis, la inhibición de la caspasa 9 redujo considerablemente la apoptosis en el recorte1Nes–PNJ KO (Fig. 2g, h, Fig. Suplementaria 4e, Fig. Suplementaria 5b). Un inhibidor de la caspasa 9, Z-LEHD-FMK, inhibió efectivamente la caspasa 9 a 1 μM y causó citotoxicidad a 20 y 40 μM (Fig. 2h, Fig. Suplementaria 4f). Para identificar una causa de aumento de la apoptosis, no detectamos un aumento en el daño del ADN (Fig. 5c–f) pero detectó fuertes aumentos de las señales de p53 en NPC positivos para SOX2 (Fig. 5g–yo). Estos datos sugieren que la recorte1Nes–El embrión KO mostró un control desregulado de la apoptosis intrínseca mediada por p53 en NPC. Proponemos que SNIP1 suprime principalmente la apoptosis intrínseca como parte de un programa de desarrollo neurológico.
genes regulados a la baja en recorte1Nes–Los NPC KO se enriquecieron en funciones relacionadas con el desarrollo del cerebro anterior y la corteza, la diferenciación de neuronas del SNC, la segregación cromosómica, la proliferación de NPC, la axonogénesis, la horquilla de replicación y las vías de señalización que involucran TLR y Rho (Fig. 2i). Aunque el recorte1Nes–Los tejidos del cerebro anterior de KO mostraron un adelgazamiento severo como consecuencia de la apoptosis, el marcador del cerebro anterior FOXG1 y el marcador del cerebro medio / posterior OTX2 se detectaron de manera similar entre el control y recorte1Nes–Cerebros KO (Fig. 5j). Estos datos sugieren que el agotamiento de SNIP1 no alteró la especificación del cerebro anterior.
Otros genes regulados a la baja en el recorte1Nes–Los NPC KO estaban involucrados en el control de la autorrenovación (Fig. 2i). Caracterización de la recorte1Nes–Los NPC KO in vitro mostraron que, en comparación con los NPC de control, cultivados recorte1Nes–Los NPC KO habían reducido la expresión de SOX2 (Fig. 6a, b). Al permitir que los NPC formen neuroesferas en suspensión y mediante pases en serie, observamos que el número de neuroesferas y el área transversal fueron significativamente más bajos en recorte1Nes–KO en comparación con el control (Fig. 6c-e). La sobreexpresión de SNIP1 humano (85% de identidad con SNIP1 de ratón) fue suficiente para rescatar la autorrenovación en cultivos recorte1Nes–NPC KO (Fig. 6f, gramo), lo que sugiere la conservación funcional de SNIP1 entre humanos y ratones. En general, concluimos que SNIP1 promueve la supervivencia celular para mantener la autorrenovación de NPC.
SNIP1 suprime la diferenciación en NPC
Debido a que en E13.5, el cerebro embrionario sufre neurogénesis, examinamos el marcador de neuronas inmaduras TUJ1. El grosor relativo de la región positiva para TUJ1 no difirió significativamente entre recorte1Nes–KO y control en E13.5 (Fig. 7a, b). Teniendo en cuenta que recorte1Nes–Los NPC KO se agotaron progresivamente, esta falta de diferencia fue una sorpresa. Por lo tanto, examinamos el efecto molecular de SNIP1 en la diferenciación de células neurales, que han perdido la expresión del marcador NPC SOX2. Realizamos RNA-seq de SOX2: células negativas para GFP clasificadas de E13.5 recorte1Nes–KO y cerebros de control de hermanos (Fig. 2a, Fig. Suplementaria 4a). Usando los criterios de fold-change >2 y p < 0.05 para comparar conjuntos de datos de 2 réplicas, cada uno de control y recorte1Nes-KO, identificamos 658 genes regulados al alza y 150 genes regulados a la baja en recorte1Nes-KO (Fig. 7c). GSEA reveló que los genes regulados al alza en recorte1Nes–Las células KO se enriquecieron en funciones relacionadas con la eliminación apoptótica, especificación y diferenciación neuronal, marcadores del mesencéfalo y promotores conocidos de alta densidad de CpG ocupados por marcas bivalentes (H3K27me3 y H3K4me3) en NPC50 (Fig. complementaria. 7d). genes regulados a la baja en recorte1Nes–Las células KO se enriquecieron en funciones relacionadas con el espliceosoma, la traducción y el ribosoma, la organización del nucleosoma y la apoptosis a través de p21 pero no de p53 (Fig. 7e). Estos resultados sugieren que SNIP1 suprime la apoptosis, la especificación y diferenciación neuronal, los programas genéticos del mesencéfalo y los genes ocupados por H3K27me3. En E13.5, aunque la apoptosis aumentada reduce recorte1Nes–NPC KO y progenitores intermedios, cuyo remanente da lugar a células que habían regulado al alza la especificación y diferenciación neuronal. Estos en combinación probablemente conducen a ninguna diferencia aparente en el grosor cortical positivo para TUJ1 entre recorte1Nes–KO y control.
A continuación, analizamos SNIP1 a través de la diferenciación in vitro de NPC. Agotamos SNIP1 en el día 1 o el día 5 mediante la transducción recorte1[flox/flox] NPC con control lentiviral o Cre y expresión génica perfilada en el día 14 (Fig. 7f). La qPCR cuantitativa mostró que, en comparación con las células de control, las células sin SNIP1 aumentaron los marcadores neuronales y gliales, pero no los marcadores NPC (Fig. 7g, h). El momento del agotamiento de SNIP1 (Día 1 frente a Día 5) no afectó este patrón de expresión génica (Fig. 7g, h). Estos resultados respaldan aún más que SNIP1 suprime la neurogénesis en los NPC. Como la eliminación global (KO) de Snip1 en embriones de pez cebra provoca una reducción de las neuronas GABAérgicas y glutamatérgicas35, preguntamos si el agotamiento de SNIP1 altera la especificación del linaje subneuronal en ratones. Debido a la pérdida drástica de tejido cerebral en recorte1Nes–KO, no pudimos analizar sólidamente el desarrollo del cerebro más allá de E13.5. En E13.5, los niveles de transcripción de marcadores neuronales GABAérgicos gad1 y Slc6a1 fueron más bajos en recorte1Nes–KO, y los marcadores neuronales glutamatérgicos no difirieron entre recorte1Nes–KO y control (Fig. 7i,j). La cuantificación de la inmunofluorescencia mostró células positivas GABA- (marcador neuronal GABAérgico) significativamente más bajas en recorte1Nes-KO (Fig. 7k, yo), lo que sugiere que la participación de SNIP1 en la especificación de linajes subneuronales puede conservarse en especies de orden superior.
SNIP1 regula directamente genes con modificaciones H3K27
Para determinar si las proteínas SNIP1 se unen directamente a los loci de genes para regular su expresión, perfilamos la distribución de SNIP1 en todo el genoma mediante CUT&RUN51 in recorte1Nes-KO y control de NPCs. Usando SICER52 y MACS253 con FDR < 0.05 para comparar 2 conjuntos de datos cada uno de control y recorte1Nes–KO, identificamos 23,188 regiones vinculadas a SNIP1 en NPC de control y solo 4187 regiones en recorte1Nes–NPC KO (Fig. 8a – c). Las 4187 regiones son el 18% de las que están en control y son una consecuencia de las proteínas SNIP1 remanentes en recorte1Nes–PNJ KO (Fig. 1a). Los mapas de calor mostraron una reducción drástica de las señales SNIP1 CUT&RUN en recorte1Nes-KO, lo que sugiere la alta especificidad de SNIP1 CUT&RUN en NPC (Fig. 3a). Aproximadamente el 50 % de los picos unidos a SNIP1 estaban dentro de los promotores (dentro de 2 kb de los sitios de inicio de la transcripción), el 7.4 % estaban ubicados en los exones, el 23.3 % en los intrones, el 0.7 % en los sitios de terminación de la transcripción, el 9.7 % en los 5′ distales (2–50 kb de un gen), 3.4 % en regiones 3′ distales (2–50 kb de un gen) y 5.5 % en regiones intergénicas (más de 50 kb de un gen) (Fig. 8d). Solo el 18.6% de los picos unidos a SNIP1 se ubicaron distales a un gen (Fig. 8d).
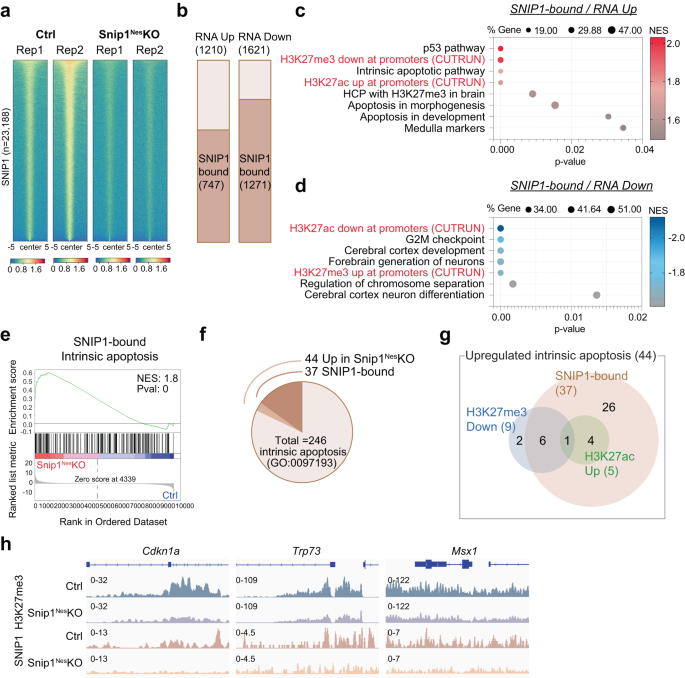
a Mapas de calor que representan la intensidad de unión de 2 réplicas biológicas de SNIP1 CUT&RUN en control y recorte1Nes–NPC KO. Se muestra la intensidad de enlace para 5 kb a cada lado de los 23,188 1 picos de CORTE Y EJECUCIÓN de SNIPXNUMX. El azul indica baja intensidad y el rojo indica alta intensidad. b Gráficos de barras que muestran el número de genes regulados al alza y a la baja (recorte1Nes–KO frente a control, FDR <0.05) que están unidos por SNIP1 en su cuerpo genético. c, d Gráficos de burbujas de los conjuntos de genes enriquecidos en genes unidos a SNIP1 que se convirtieron en genes regulados al alza y genes regulados a la baja en recorte1Nes-KO contra NPC de control. Al agregar nuestros datos H3K27me3/ac CUT&RUN a los conjuntos de genes GSEA, los niveles de H3K27me3 y H3K27ac mostraron anticorrelación y correlación con la expresión génica, respectivamente. Los datos de origen se proporcionan en un archivo de datos de origen. e GSEA representativo de genes regulados al alza en recorte1Nes-KO NPC frente a NPC de control que están vinculados por SNIP1 en NPC de control. Los genes de apoptosis intrínsecos se unieron principalmente a SNIP1 y se enriquecieron en los genes regulados al alza en recorte1Nes-KO NPC. Los genes expresados diferencialmente se clasificaron primero por su cambio de pliegue y p-valor antes de que se realizara GSEA. f Gráfico circular que muestra las proporciones de genes de apoptosis intrínsecos que están regulados al alza en recorte1Nes-KO NPCs y/o obligado por SNIP1. g Diagrama de Venn que muestra el número de genes de apoptosis intrínseca regulados al alza en 3 categorías. Usando nuestros datos SNIP1, H3K27me3 y H3K27ac CUT&RUN, los 44 genes se clasificaron en 1) SNIP1 enlazado en NPC de control, 2) niveles reducidos de H3K27me3 en recorte1Nes-KO contra PNJ de control (p < 0.05), y/o 3) aumento de los niveles de H3K27ac en recorte1Nes-KO contra PNJ de control (p <0.05). h Pistas H3K27me3 y SNIP1 CUT&RUN visualizadas por Integrative Genomics Viewer (IGV) en genes de apoptosis intrínseca regulados al alza. cdkn1a, Chr17: 29,090,888 29,095,850 XNUMX − XNUMX XNUMX XNUMX. Trp73, Chr4: 154,132,565 154,143,373 XNUMX − XNUMX XNUMX XNUMX. msx1, Chr5: 37,818,429 37,828,924 XNUMX − XNUMX XNUMX XNUMX.
A continuación, examinamos si la ocupación de SNIP1 se correlaciona con la expresión génica. De los 1210 genes regulados al alza y los 1621 genes regulados a la baja en recorte1Nes-KO, 747 (62%) y 1,271 (78%) fueron objetivos SNIP1, respectivamente, en los NPC de control (Fig. 3b). Estas superposiciones son significativas, con p = 5.55e-25 y 2.24e-155, respectivamente, por la prueba hipergeométrica (dado un total de 21,636 genes expresados), lo que sugiere que SNIP1 ocupa estos genes para regular su expresión. GSEA mostró que los objetivos SNIP1 que se regularon al alza en recorte1Nes–Los KO se enriquecieron en la vía p53, médula/mesencéfalo y apoptosis (Fig. 3c), mientras que los objetivos SNIP1 que se regularon a la baja en recorte1Nes–Los KO estaban enriquecidos en el punto de control G2/M, desarrollo cortical y segregación cromosómica (Fig. 3d). De los 1,621 genes regulados a la baja en recorte1Nes–KO, 1,093 se unieron a SNIP1 (no a PRC2) y se enriquecieron en programas genéticos en el punto de control G2-M, huso mitótico, vías de señalización clave para el neurodesarrollo y apoptosis (Fig. 8e), lo que sugiere que SNIP1 promueve su expresión. Los genes de apoptosis intrínsecos se enriquecieron en objetivos SNIP1 que se regularon positivamente en recorte1Nes-KO (fig. 3e,f, Fig. Suplementaria 8f, gramo). De los 44 genes en el conjunto de genes de apoptosis intrínsecos, 37 promotores se unieron a SNIP1 en NPC de control (p = 4.62e-5; Higo. 3g), lo que sugiere que SNIP1 suprime directamente estos genes. Estos datos sugieren que SNIP1 regula directamente los programas genéticos cruciales para la apoptosis y el ciclo celular.
Genes regulados al alza en recorte1Nes-KO se enriquecieron en genes cuyos promotores de alta densidad de CpG que están 1) ocupados por H3K27me3 en el cerebro murino embrionario50 o 2) bivalente en NPC de ratón54 (Fig. complementaria. 9a, b). Esto nos llevó a analizar si SNIP1 controla los programas genéticos a través de modificaciones H3K27. Perfilamos H3K27me3 y H3K27ac de CUT&RUN en recorte1Nes-KO y NPC positivos para SOX2 de control (Fig. 9c-g). Observamos una fuerte correlación entre los genes regulados al alza, la ocupación más baja de H3K27me3 y la ocupación más alta de H3K27ac, mientras que los genes regulados a la baja tenían una ocupación más alta de H3K27me3 y una ocupación más baja de H3K27ac en recorte1Nes-KO (fig. 3c, d, Fig. Suplementaria 9h, yo). Entre los 44 genes de apoptosis intrínseca regulados al alza, 9 genes mostraron una ocupación reducida de H3K27me3 y 5 genes mostraron un aumento en la ocupación de H3K27ac.p < 0.05, Fig. 3g, h, Fig. Suplementaria 9j,k). Estos datos sugieren que SNIP1 en la cromatina controla las modificaciones de H3K27 y la expresión génica.
Las vías de señalización de TGFβ y NFκB controlan la localización de SNIP1 en la cromatina
Como SNIP1 participa en TGFβ22,23,24,25 y NFκB26,27,28,29,30 vías de señalización, nuestro objetivo era probar si su inhibición afecta la apoptosis en los NPC agotados de SNIP1. Probamos 3 inhibidores de la señalización de TGFβ o NFκB en busca de toxicidad para los NPC (Fig. 4a, b). En concentraciones de 0.1 a 0.5 μM, K02288 dirigido a TGFβ (pero no a Galunisertib o LDN-193189) redujo consistentemente la apoptosis en los NPC agotados de SNIP1 (Fig. 4c-e). Por el contrario, 2 inhibidores de la señalización de TGFβ y 3 inhibidores de la señalización de NFκB aumentaron la apoptosis en los NPC (Fig. 4f-h). Por lo tanto, decidimos probar si alguno de los 6 inhibidores altera la unión de SNIP1 a la cromatina. Tratamos los NPC con DMSO o inhibidores a 0.5 μM durante 3 días y realizamos CUT&RUN para analizar la localización de SNIP1 en la cromatina (Fig. 4i). Analizando 2 datos de CUT&RUN SNIP1 replicados por condición de tratamiento, usamos p < 0.05 para identificar diferencias significativas y constantes en las señales SNIP1 CUT&RUN entre los tratamientos con inhibidores y de control. Identificamos los cambios de SNIP1 CUT&RUN inducidos por el tratamiento con K02288 (Datos complementarios 1). También identificamos cambios significativos en SNIP1 CUT&RUN en genes unidos a SNIP1 que se superponen en cualquiera de los 2 tratamientos con inhibidores (Datos complementarios 1). El perfil promedio de las señales SNIP1 CUT&RUN en los promotores unidos a SNIP1 en NPC tratados con diferentes inhibidores confirmó que la inhibición de TGFβ (Fig. 4j,k) y NFκB (Fig. 4l, metro) señalización alterada significativamente la unión de SNIP1 a los promotores. Estos datos sugieren que las vías de señalización de TGFβ y NFκB controlan la unión de SNIP1 a loci de genes específicos en NPC.
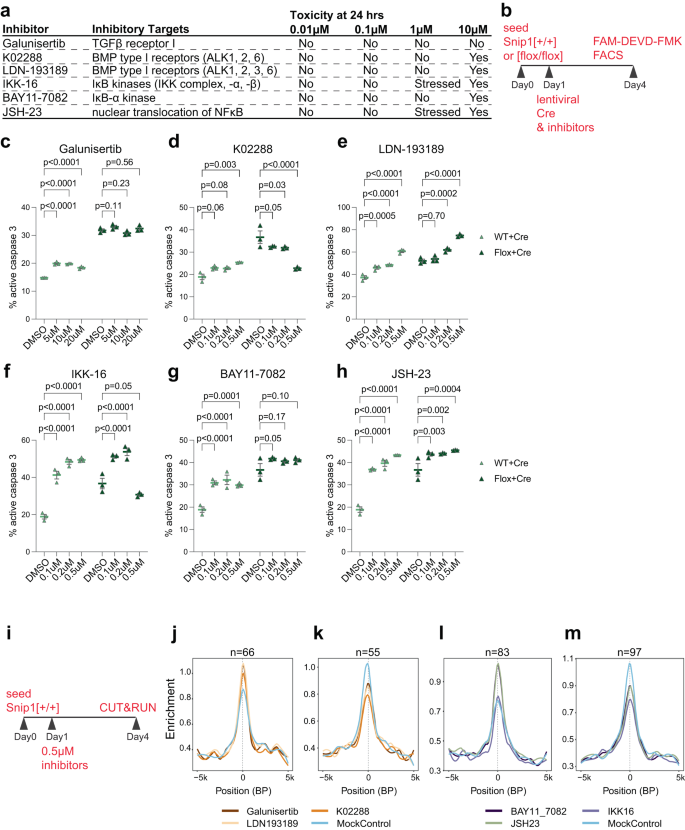
a Inhibidores dirigidos a componentes en las vías de señalización de TGFβ y NFκB y su citotoxicidad a diferentes concentraciones. b Esquema del ensayo de inhibidor. El día 1, WT o recorte1Los NPC [flox/flox] se trataron con inhibidor o DMSO y se transdujeron con Cre lentiviral para el agotamiento de SNIP1. FAM-DEVD-FMK se utilizó para analizar cl-caspasa 3 mediante FACS el día 4. c-h El porcentaje de células con caspasa 3 activa cuantificado por FACS. Se agregaron inhibidores a diferentes concentraciones junto con mCherry-Cre lentivirus. Se muestra el porcentaje de FAM-FLICA (caspasa 3 activa)-/mCherry-doble población positiva (fuera de la población mCherry-positiva). N = 3 para cada tratamiento. Los datos se presentan como media ± SEM, y se utilizó ANOVA de dos vías para el análisis estadístico. Los gráficos FACS representativos se muestran en la figura complementaria. 14c, d. Los datos de origen se proporcionan en un archivo de datos de origen. i Esquema de SNIP1 CUT&RUN con tratamiento inhibidor. En el día 1, los NPC se trataron con control de DMSO o diferentes inhibidores. Se realizó una réplica de SNIP1 CUT&RUN para cada uno de los 7 tratamientos en el día 4. j-m Gráficos de perfil que comparan la mediana de la intensidad de unión de SNIP1 en NPC en los objetivos unidos a SNIP1 que tenían una unión de SNIP1 significativamente más alta o más baja en los inhibidores versus el tratamiento de control con DMSO. n indica números de región. Las regiones se consideraron verdaderos objetivos de SNIP1 cuando los niveles de SNIP1 se redujeron en recorte1Nes-KO contra PNJs de control con p <0.05.
PRC2 requiere SNIP1 para la localización en cromatina y depósito de H3K27me3
Investigamos las interacciones entre SNIP1, H3K27 metiltransferasa PRC2 e histona acetiltransferasas p300 y CBP. Anticuerpo anti-SNIP1 co-inmunoprecipitado con subunidades PRC2 conocidas Jumonji y dominio de interacción rico en AT que contiene 2 (JARID2), supresor de zeste 12 (SUZ12) y EZH2, pero no el control negativo RBBP5 en el extracto nuclear de NPC (Fig. 5a). El anticuerpo anti-SNIP1 no se co-inmunoprecipitó con las subunidades PRC2 en los NPC agotados de SNIP1 (Fig. 10a), apoyando la especificidad del anticuerpo. El anticuerpo anti-JARID2, EZH2 o EED co-inmunoprecipitó SNIP1 y otras subunidades de PRC2, pero no el control negativo RBBP5 en el extracto nuclear de NPC (Fig. 5b–d). El anticuerpo anti-p300 o CBP no logró co-inmunoprecipitar SNIP1, lo que sugiere que en los NPC, su interacción física es indetectable (Fig. 10b, c). La unión de PRC2-SNIP1 está respaldada por otros estudios de proteómica que mostraron que PRC2 co-inmunoprecipita SNIP119,20.
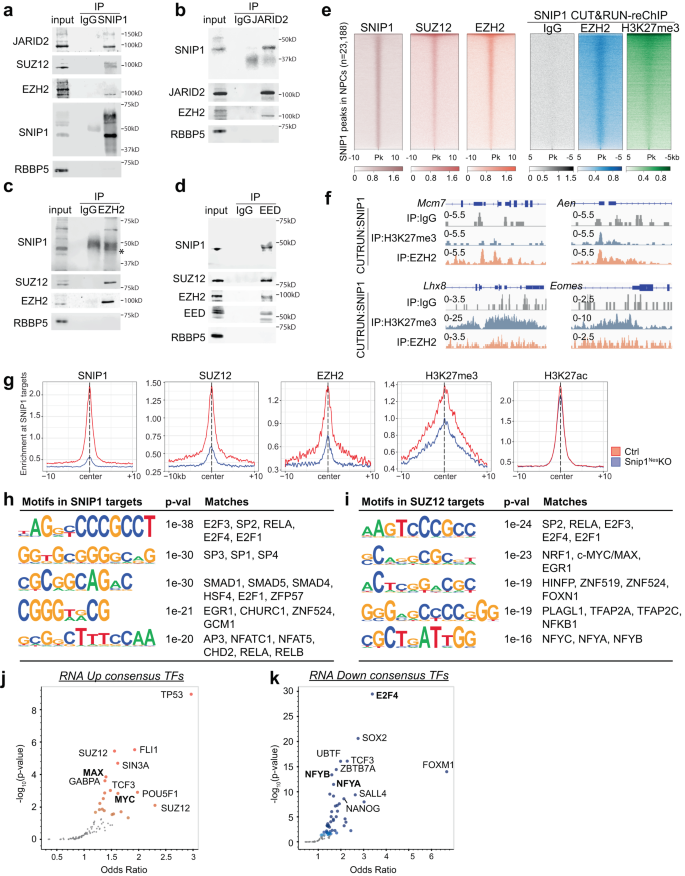
anuncio Co-inmunoprecipitación seguida de WB para examinar la interacción entre SNIP1 y PRC2. (a) SNIP1, (b) JARID2, (c) EZH2, o (d) Se inmunoprecipitó EED en el extracto nuclear de NPC. RBBP5 fue un control negativo. Los datos de origen se proporcionan como un archivo de datos de origen. e Mapas de calor que alinean los picos de cromatina enriquecidos con SNIP1, SUZ12 y EZH2 en NPC. Los picos de SNIP1 CUT&RUN–reChIP con IgG, EZH2 y H3K27me3 se alinearon con los picos unidos a SNIP1. Se muestra la intensidad para 5 o 10 kb a ambos lados de 23,188 picos unidos a SNIP1. Un color oscuro indica alta intensidad y un color claro indica baja intensidad. f Las pistas de SNIP1 CUT&RUN–reChIP con IgG, EZH2 y H3K27me3 se visualizan mediante Integrative Genomics Viewer (IGV). mcm7, Chr5: 138,169,717 − 138,173,621. Aen, Chr7: 78,894,526 − 78,898,271. Lhx8, Chr3: 154,325,066 − 154,334,835. Eomas, Chr9: 118,474,178 − 118,480,775. g Gráficos de perfil que comparan la intensidad de unión media de SNIP1, PRC2 y H3K27me3/ac en recorte1Nes-KO contra NPC de control en los objetivos SNIP1. Las regiones se consideraron verdaderos objetivos de SNIP1 cuando los niveles de SNIP1 se redujeron en recorte1Nes-KO contra PNJs de control con p <0.05. h, i Motivos de regiones vinculadas a SNIP1 y SUZ12 donde sus niveles se redujeron significativamente en recorte1Nes–NPC KO con cambio de pliegue> 2 y p <0.05. Se realizó un análisis HOMER de novo y los cinco motivos con menor p-los valores y las coincidencias de motivos de vertebrados se enumeran aquí. j, k Gráficos de volcanes de factores de transcripción cuya unión a nuestros genes expresados diferencialmente (FDR <0.05) ha sido reportada. Los genes se buscaron contra ENCODE y ChEA consenso TF de la base de datos ChIP-X usando Enrichr56. Los colores más oscuros muestran más pequeños p-valores y puntos grandes pasados p-valor < 0.05. Los factores de transcripción en negrita se encontraron en nuestros análisis de motivos CUT&RUN en la Fig. 5h, i.
Probamos si SNIP1 altera la expresión de las subunidades PRC2. Aunque el agotamiento de SNIP1 disminuyó Ezh2 niveles de transcripción, no alteró los niveles de proteína de las subunidades PRC2, H3K27me3 o H3K27ac (Fig. 10d-g). A continuación, realizamos CUT&RUN para perfilar SUZ12 y EZH2 en recorte1Nes-KO y NPC de control (Fig. 10 h – j). De acuerdo con los resultados de co-inmunoprecipitación, SUZ12 y EZH2 coocupan los sitios objetivo unidos a SNIP1 en todo el genoma (Fig. 5e). Para analizar las interacciones PRC2-SNIP1 en la cromatina, realizamos CUT&RUN-reChIP. En este ensayo, la cromatina liberada por SNIP1 CUT&RUN fue inmunoprecipitada por IgG, EZH2 o H3K27me3. Los lugares representativos mcm7, Aen, Lhx8y Eomas tenía co-ocupación de SNIP1 con EZH2 y H3K27me3 pero no control negativo IgG (Fig. 5f). En todo el genoma, CUT&RUN-reChIP mostró que EZH2 y H3K27me3 tenían grandes superposiciones con SNIP1 y PRC2 en los picos unidos a SNIP1 (Fig. 5e).
Al examinar el papel de SNIP1 en la ocupación de cromatina PRC2, encontramos que los niveles de SUZ12, EZH2 y H3K27me3 en la cromatina se redujeron significativamente en recorte1Nes-KO (fig. 5g, figuras complementarias. 10k, 11a). Por el contrario, los niveles de H3K27ac estaban menos alterados en recorte1Nes-KO NPC (Fig. 5g, figuras complementarias. 10k, 11a). A continuación, utilizamos un ensayo in vitro para analizar el efecto cinético del agotamiento de SNIP1 en PRC2. En comparación con el lentivirus de control, la transducción lentiviral Cre de recorte1Los NPC [flox/flox] agotaron las transcripciones de SNIP1 en un 70 % y un 99.9 % en el segundo y tercer día, respectivamente. Esto no alteró el nivel de transcripción de los componentes de PRC2 (Fig. 11b). Usando EZH2 y SUZ12 CUT&RUN, observamos una fuerte reducción de PRC2 en la cromatina al tercer día del agotamiento de SNIP1 (Fig. 11c). Juntos, estos datos respaldan que PRC2 requiere SNIP1 para unirse a la cromatina.
Para caracterizar los objetivos SNIP1-PRC2, realizamos el descubrimiento de motivos de novo mediante el software HOMER55. Usando los criterios de fold-change >2 y p < 0.05 en control vs recorte1Nes-KO, los objetivos SNIP1 se enriquecieron en motivos de proteínas E2F, SP1/3/4 y EGR1 (Fig. 5h). Los motivos de los interactores SNIP1 informados anteriormente, las proteínas SMAD y RELA, también se encontraron entre los objetivos de SNIP121,26. En los picos unidos a SUZ12 que habían reducido la unión en recorte1Nes-KO, los motivos se enriquecieron con las subunidades SP2, RELA, proteínas E2F, EGR1, HINFP, PLAGL1 y NF-Y (Fig. 5i). Las similitudes de los motivos unidos a SNIP1 y SUZ12 apuntan a posibles interacciones de SNIP1 y PRC2 con algunos de estos factores de transcripción.
A continuación, examinamos si los motivos unidos a SNIP1 o SUZ12 estaban sobrerrepresentados en genes expresados diferencialmente. Usando Enriquecer56, encontramos que los genes regulados al alza en recorte1Nes-KO fueron objetivos de TP53, FLI1, SUZ12, MAX y MYC, mientras que los genes regulados a la baja fueron objetivos de E2F4, SOX2, NFYB y NFYA (Fig. 5j,k). Las proteínas E2F se descubrieron mediante análisis de motivo y Enrichr, y los objetivos E2F4 mcm7 y Anp32e eran objetivos SNIP1 y PRC2 que habían reducido la unión en recorte1Nes-KO (fig. 5k, Fig. Suplementaria 11d). Entre los genes regulados al alza en recorte1Nes-Los objetivos KO, MYC y MAX se identificaron mediante análisis de motivo y Enrichr (Fig. 5j, Fig. Suplementaria 11e). Estos datos sugieren que las proteínas E2F, MYC y MAX pueden influir en las actividades de SNIP1-PRC2 para la regulación génica.
PRC2 promueve la apoptosis en ausencia de SNIP1
Probamos el papel de PRC2 en la supervivencia de NPC in vivo. Nosotros usamos Nes::Cre para extirpar los exones 3 a 6 de eed (una subunidad central PRC2) para generar eedNes-KO y recorte1Nes-EedNes-dKO (Fig. 12a, b). El agotamiento de EED en NPC no indujo apoptosis en eedNes-KO E13.5 cerebro (Fig. 12c-g). Otros habían estudiado EED en el control y eedemx1-KO telencéfalo dorsal y sacamos conclusiones57 que difería de nuestras observaciones en eedNes-KO E13.5 cerebro. Esta diferencia podría explicarse por la expresión de emx1::Cre y Nes::Cre en diferentes etapas de desarrollo y regiones del cerebro. Se observó apoptosis en otras regiones cerebrales de control y eedNes-KO cerebros (Fig. 12c, d). Como ya se ha demostrado que EED afecta la neurogénesis en E14.5 pero no en E16.557, el efecto exacto de EED/PRC2 en las funciones de NPC depende de la etapa de desarrollo y del contexto celular. Nuestro análisis mostró que, en comparación con recorte1Nes–KO recorte1Nes-EedNes-dKO tenía significativamente menos células positivas para cl-caspasa 3 (Fig. 6a – c), más NPC SOX2 positivos (Fig. 6d, e), y más progenitores intermedios positivos para TBR2 o INSM1 (Fig. 6f–yo). Las neuronas inmaduras positivas para TUJ1 no se vieron marcadamente afectadas por la interacción funcional SNIP1-PRC2 (Fig. 12h, yo). Agotamiento de EED en el recorte1Nes–El cerebro embrionario KO redujo la apoptosis y rescató NPC y progenitores intermedios. Por lo tanto, SNIP1 suprime la apoptosis en el cerebro en desarrollo al contrarrestar a PRC2.
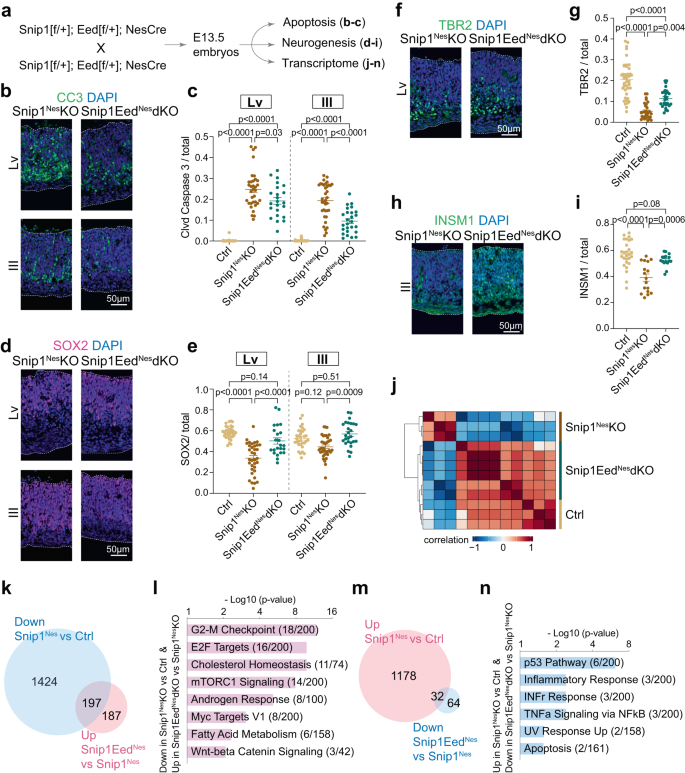
a Esquema del cruce genético que agota tanto SNIP1 como EED para ensayos posteriores. b, c Análisis IF de CC3 superpuesto con DAPI del cerebro E13.5. Barra, 50 μm. Cada punto de datos representa una imagen. Ocho embriones de control, 7 recorte1Nes-KO embriones, y 5 recorte1Nes-EedNes-Se analizaron embriones dKO. Para el ventrículo lateral, n = 38 imágenes (control), n = 34 (recorte1Nes-KO), y n = 23 (recorte1Nes-EedNes-dKO). Para el tercer ventrículo, n = 38 (testigo), n = 36 (recorte1Nes-KO), y n = 26 (recorte1Nes-EedNes-dKO). d-yo IF del marcador NPC SOX2 y marcadores progenitores intermedios TBR2 e INSM1 superpuestos con DAPI del cerebro E13.5. Barra, 50 μm. Las poblaciones de (e) SOX2 positivo, (g) TBR2 positivo, o (i) Se cuantificaron las células positivas para INSM1 en el revestimiento neuroepitelial de los ventrículos lateral y/o tercero. Cada punto de datos representa una imagen. En (g), n = 43 imágenes (control), n = 30 (recorte1Nes-KO), y n = 29 (recorte1Nes-EedNes-dKO). En (i), n = 27 (testigo), n = 17 (recorte1Nes-KO), y n = 16 (recorte1Nes-EedNes-dKO). Para Paneles c, e, gy i, los datos se presentan como media ± SEM, y se utilizó ANOVA de dos vías para el análisis estadístico. j Agrupación no supervisada de datos de RNA-seq del control (n = 3), recorte1Nes-KO (n = 3), y recorte1Nes-EedNes-dKO (n = 6) cerebros en E13.5. Los ARN de las regiones del prosencéfalo y el mesencéfalo se secuenciaron y fusionaron para análisis posteriores. El azul indica una correlación negativa y el rojo indica una correlación positiva. k, m Diagramas de Venn que muestran el número de genes expresados diferencialmente con FDR <0.05. Las listas de genes regulados a la baja en recorte1Nes-KO vs control y genes regulados al alza en recorte1Nes-EedNes-dKO vs. recorte1Nes-KO se comparan en (k). Las listas de genes sobrerregulados en recorte1Nes-KO vs control y genes regulados a la baja en recorte1Nes-EedNes-dKO vs. recorte1Nes-KO se comparan en (m). l, n Ontología génica de los genes rescatados correspondientes a la Fig. 6k, metro. Los genes se buscaron en la base de datos de firmas moleculares (MSigDB) Hallmark 2020 usando Enrichr56. Los datos de origen se proporcionan en un archivo de datos de origen (c, e, g, i, l, n).
Perfilamos los transcriptomas de control, recorte1Nes–KO, y recorte1Nes-EedNes-dKO tejidos cerebrales. El agrupamiento no supervisado basado en los 3,000 genes más expresados diferencialmente (basado en valores de variación medianos) sugiere una mayor similitud en los transcriptomas entre el control y recorte1Nes-EedNes-dKO en comparación con recorte1Nes–KO (fig. 6j). Usando fold-change >2 y p < 0.05 para comparar conjuntos de datos de recorte1Nes-KO y recorte1Nes-EedNes-dKO, identificamos 184 genes regulados al alza y 994 genes regulados a la baja en recorte1Nes-EedNes-dKO (Fig. 13a).
Proporcionar una posible explicación molecular del rescate de NPC y progenitores intermedios en recorte1Nes-EedNes-dKO, analizamos genes expresados diferencialmente entre el control, recorte1Nes-EedNes-dKO, y recorte1Nes-KO. Encontramos que 197 genes regulados a la baja en recorte1Nes–KO recuperó parcialmente la expresión en recorte1Nes-EedNes-dKO (Fig. 6k). Estos genes rescatados están involucrados en el punto de control G2/M, los objetivos E2F, la homeostasis del colesterol, la señalización de mTORC1 y la respuesta a los andrógenos (Fig. 6l). Por el contrario, 32 genes regulados al alza en recorte1Nes-KO se reguló a la baja en recorte1Nes-EedNes-KO (Higo. 6m) y están involucrados en la vía p53, las respuestas inflamatorias y de interferón gamma, la señalización de NFκB y la apoptosis (Fig. 6n). De estos 32 genes, cdkn1a es el único gen de apoptosis intrínseco con niveles significativamente más bajos de H3K27me3 en recorte1Nes-KO versus control (FDR < 0.05; Fig. 13b), lo que sugiere que SNIP1 promueve directamente PRC2 y H3K27me3 en el cdkn1a lugar. Nuestro perfil de ocupación del genoma SNIP1-PRC2 sugiere que SNIP1 inhibe la deposición de H3K27me3 en loci que incluyen objetivos E2F (Fig. 11d) pero promueve la deposición de H3K27me3 en otros loci que incluyen genes apoptóticos cdkn1a, Nkx2-9, etv4, pxdc1y Tap1 (Fig. complementaria. 13b, c). En conjunto, SNIP1 ejerce un control dependiente de loci de la deposición de H3K27me3 y la expresión génica para equilibrar la división, la apoptosis y la diferenciación de NPC en el cerebro en desarrollo.
- Distribución de relaciones públicas y contenido potenciado por SEO. Consiga amplificado hoy.
- PlatoData.Network Vertical Generativo Ai. Empodérate. Accede Aquí.
- PlatoAiStream. Inteligencia Web3. Conocimiento amplificado. Accede Aquí.
- PlatoESG. Automoción / vehículos eléctricos, Carbón, tecnología limpia, Energía, Ambiente, Solar, Gestión de residuos. Accede Aquí.
- Desplazamientos de bloque. Modernización de la propiedad de compensaciones ambientales. Accede Aquí.
- Fuente: https://www.nature.com/articles/s41467-023-40487-4

