
Este artículo destaca algunas de las similitudes y diferencias entre las vías regulatorias de Dispositivos Médicos 510(k) y el Marcado CE y ayuda a armonizar algunos aspectos de una estrategia regulatoria general.
Nota del editor: este artículo actualiza y reemplaza el blog de 2014 de Vincent Crabtree sobre 510(k) y el marcado CE (partes 1 y 2).
La FDA tiene una definición clara de la vía 510(k) que permite a los fabricantes llevar sus productos al mercado a un ritmo más rápido y a menor costo en comparación con la vía de aprobación previa a la comercialización (PMA) de la FDA. El proceso de marcado CE de la Unión Europea, que originalmente se consideraba sencillo si el dispositivo estaba aprobado por la FDA, ya no es un proceso trivial. La introducción del MDR (2017/745) y el IVDR (2017/746) de la UE ha llevado a varios fabricantes a iniciar programas de remediación para actualizar sus documentos de cumplimiento y realizar pruebas o estudios clínicos adicionales.
Resumen Ejecutivo
Las Directivas de Dispositivos Médicos (MDD) están en transición a las Regulaciones de Dispositivos Médicos (MDR) en la UE, que reclasifican los dispositivos médicos en Clase I, Is, Im, Ir, IIa, IIb, III y IIIc (personalizados). La clasificación del dispositivo orienta los requisitos de diseño, prueba, verificación, validación, clínica y vigilancia posterior a la comercialización para que el dispositivo obtenga la marca CE y se coloque en el mercado de la UE.
El proceso 510(k) y el proceso de Marcado CE no se pueden comparar uno a uno porque el proceso 510(k) se aplica a una cantidad relativamente pequeña de productos en comparación con la aplicabilidad del proceso de Marcado CE. Los cambios significativos en los dispositivos requieren cierto nivel de monitoreo e informes en ambos procesos y pueden llevar a la necesidad de un nuevo 510(k) o una nueva inspección por parte de los Organismos Notificados.
El proceso 510(k) ha evolucionado para ser sencillo con el programa eSTAR y la funcionalidad de seguimiento del progreso. El Actualmente se está poniendo a prueba el programa eSTAR para un portal conjunto de la FDA y Health Canada., lo que hará que el proceso sea más conveniente para los fabricantes. EUDAMED se encuentra actualmente en construcción y se espera que un sistema funcional completo esté disponible en el tercer trimestre de 3.
Tanto el proceso 510(k) como el de Marcado CE operan bajo diferentes conjuntos de regulaciones para el Sistema de Gestión de Calidad, pero ISO 13485:2016 se puede emplear con una implementación adicional de las cláusulas FDA y MDR de la UE. Con la propuesta de armonizar QSR con ISO 13485:2016, la implementación de un Sistema de Gestión de Calidad será más fácil para los fabricantes. Se requiere documentación básica de diseño y gestión de riesgos para la presentación, y la documentación complementaria varía según el mercado.
Terminología, equivalencia y aplicación
510(k) es una presentación previa a la comercialización realizada a la FDA para demostrar que el dispositivo que se comercializará es tan seguro y eficaz, es decir, sustancialmente equivalente (SE), como un dispositivo comercializado legalmente. La FDA define los dispositivos comercializados legalmente a los que se les asigna equivalencia como el "predicado". Un dispositivo es sustancialmente equivalente a un predicado si el dispositivo sujeto:
- tiene el mismo uso previsto que el predicado y tiene las mismas características tecnológicas o diferentes características tecnológicas que no plantean diferentes cuestiones de seguridad y eficacia.
- La información presentada a la FDA demuestra que el dispositivo es tan (o más) seguro y eficaz como el dispositivo comercializado legalmente.
Consulte la Figura 1: Equivalencia sustancial según la FDA para presentaciones 510(k).
Las presentaciones 510(k) son aplicables a dispositivos de Clase I, II (a menos que estén exentos) y dispositivos de Clase III, si corresponde.
Aunque cada país de la Unión Europea, normalmente denominados Estados miembros, tiene una autoridad competente que es responsable del cumplimiento del dispositivo relacionado con las directivas de marcado CE, la responsabilidad del marcado CE se delega a los organismos notificados, para evitar conflictos de intereses. y armonizar los requisitos.
Para obtener una marca CE para el dispositivo, el fabricante debe demostrar que su dispositivo cumple con los requisitos MDR de la UE. Normalmente, los organismos notificados tienen la tarea de revisar el registro maestro del dispositivo y los documentos asociados para aprobar el marcado CE del dispositivo. Una de las formas de demostrar el cumplimiento es mediante la equivalencia. El marcado CE, al reclamar equivalencia según el MDR de la UE, requiere consideraciones adicionales para demostrar la equivalencia. El fabricante está obligado a declarar la equivalencia de las siguientes características:
- Técnico: condiciones de uso, especificaciones y propiedades, métodos de implementación (cuando corresponda), principios de operación y requisitos críticos de desempeño.
- Biológicos: materiales o sustancias en contacto con los mismos tejidos o fluidos corporales humanos, tipo y duración similares del contacto y características de liberación de las sustancias (incluidos los productos de degradación y los lixiviables).
- Clínico: condición o propósito clínico, gravedad y etapa de la enfermedad, sitio del cuerpo, población, usuario, desempeño crítico en vista del efecto clínico esperado para un propósito específico.
Demostrar equivalencia bajo el marcado CE requiere mucho más esfuerzo y, a veces, información patentada que puede no estar disponible para intentar reclamar equivalencia con productos que pertenecen a un fabricante diferente. No se requiere la aprobación del organismo notificado para el marcado CE para la Clase I según el MDR de la UE. Los dispositivos de Clase I están autocertificados según el MDR de la UE. Los dispositivos de Clase I que se suministran estériles (Is), tienen función de medición (Im) o son un instrumento quirúrgico reutilizable (Ir) están sujetos al Marcado CE.
 y marcado CE")
Figura 1: Equivalencia sustancial según la FDA para presentaciones 510(k).
Cambios en el dispositivo
Los dispositivos que sufren cambios de diseño pueden requerir una nueva presentación 510(k). Se debe presentar un nuevo 510(k) en los siguientes casos:
- Cambios realizados con la intención de afectar significativamente la seguridad o eficacia de un dispositivo.
- Cambios importantes en el etiquetado: agregar contraindicaciones, dispositivos reetiquetados como reutilizables de un solo uso, etc.
- Cambios importantes en tecnología, ingeniería y rendimiento: cambios en el mecanismo de control, principio de funcionamiento, cambios en el tipo de energía, etc.
- Cambios de materiales
- Modificaciones que conduzcan a cambios significativos en el perfil de riesgo del dispositivo.
Si bien la lista anterior parece sencilla, existen varios escenarios en los que las líneas se difuminan y es posible que el dispositivo no requiera un nuevo 510(k). La guía de la FDA Decidir cuándo presentar un 510(k) para un cambio en un dispositivo existente proporciona diagramas de flujo detallados que ayudan con la toma de decisiones para presentar un nuevo 510(k). Una guía separada analiza cuándo cambios en el software requieren un nuevo 510(k).
Según el MDR de la UE, los dispositivos que experimenten cambios significativos en una o más de las siguientes categorías deben informar el cambio al organismo notificado que certificó el dispositivo.
- finalidad prevista
- especificación de diseño o rendimiento
- ingrediente o material
- Esterilización o diseño de embalaje con impacto en la esterilización.
- software
La definición de cambio significativo puede ser ambigua. Orientación del MDCG proporciona información adicional sobre cambios significativos bajo el MDR de la UE. El organismo notificado podrá decidir volver a auditar el sistema de gestión de la calidad o la documentación técnica del fabricante, según sea necesario.
Una buena estrategia regulatoria incluirá requisitos tanto de la FDA como del MDR de la UE para garantizar que ambos se cumplan.
Proceso y Tarifas
Las presentaciones 510(k) se pueden realizar en línea utilizando el portal CDRH. El portal CDRH permite a los fabricantes enviar un envío de dispositivos médicos mediante eSTAR, un formulario PDF interactivo. El portal CDRH también proporciona un rastreador de progreso que muestra el estado del envío. El portal tiene algunas limitaciones con respecto al tamaño y tipo de archivos, pero permite al corresponsal enviar documentos de gran tamaño al Centro de Control de Documentos (DCC) del CDRH. Cualquier dispositivo al que se le haya concedido la autorización 510(k) puede comercializarse inmediatamente mientras se espera la inspección del Sistema de Calidad de la FDA (21 CFR 820), que puede tener lugar después de la autorización.
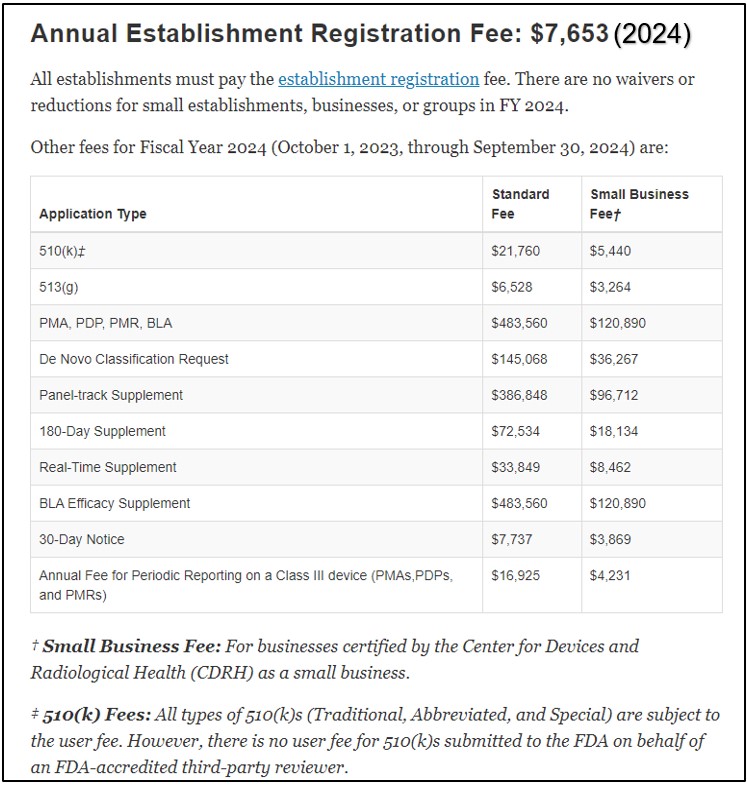
Los costos de solicitud para 510(k) se publican en el sitio web de la FDA en Enmiendas a las tarifas de usuario de dispositivos médicos (MDUFA). Las tarifas varían para empresas estándar y pequeñas empresas. La siguiente tabla proporciona una idea de la estructura de tarifas. Las tarifas se actualizan cada año fiscal. Todo establecimiento está obligado a pagar una cuota anual. tasa de registro de establecimiento.
El proceso de marcado CE requiere la participación de organismos notificados y una auditoría de SGC calificada antes de que un dispositivo pueda ser evaluado para obtener un marcado CE. Elija su organismo notificado en función de la combinación adecuada de tarifas, arreglos de viaje y experiencia que funcione para usted.
El MDR de la UE introdujo el concepto de una base de datos europea sobre dispositivos médicos (EUDAMED) que tiene como objetivo proporcionar una "imagen viva del ciclo de vida de los dispositivos médicos que están disponibles en la Unión Europea (UE)". EUDAMED estará compuesto por seis módulos relacionados con: registro de actores, identificación única de dispositivo (UDI) y registro de dispositivos, Organismos Notificados y certificados, investigaciones clínicas y estudios de desempeño, vigilancia y seguimiento del mercado. A partir de marzo de 2024, EUDAMED cuenta con tres módulos activos: operadores económicos, dispositivos y certificados. Está previsto que los tres últimos módulos estén online en el tercer trimestre de 3.
En ausencia de un sistema en línea para gestionar las solicitudes de marcado CE, el fabricante debe utilizar portales/carpetas compartidas/otros métodos establecidos por los organismos notificados para compartir la documentación y las pruebas necesarias para la certificación. El organismo notificado lleva a cabo una auditoría del sistema de gestión de calidad según los requisitos del sistema de gestión de calidad MDR de la UE (Anexo IX) antes de certificar el dispositivo para el mercado. Los dispositivos no pueden recibir la marca CE ni comercializarse sin una auditoría del SGC del organismo notificado y una revisión de la documentación técnica (o un muestreo en algunos casos), a menos que el dispositivo esté clasificado como Clase I.
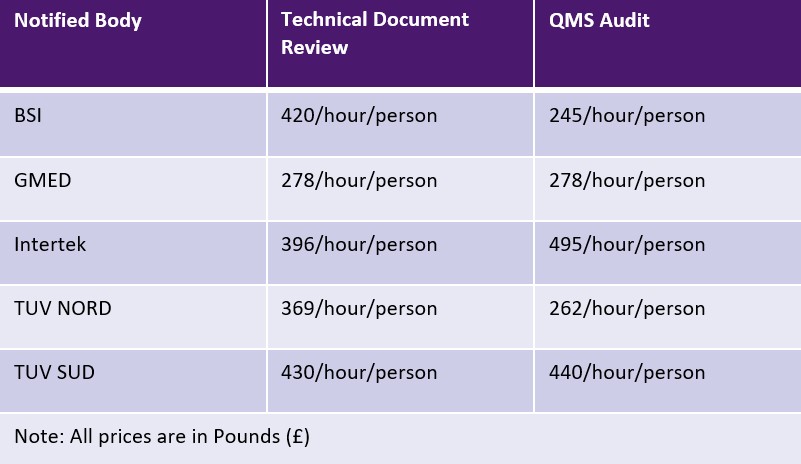
El MDR de la UE permite a los organismos notificados cobrar una tarifa fija o una tarifa basada en el tiempo para cada actividad. Sin embargo, los organismos notificados no están sujetos a ningún límite en materia de tarifas. La siguiente tabla muestra una muestra de organismos notificados y sus tarifas. Es posible que se apliquen tarifas adicionales por viajes y experiencia.
Sistema de Gestión de Calidad
Los fabricantes deben realizar su debida diligencia y garantizar que exista el sistema de gestión de calidad adecuado antes de comercializar sus dispositivos.
La FDA rige el sistema de gestión de calidad según las Regulaciones del sistema de calidad (QSR) (21 CFR 820), que se aplica a los fabricantes que pretenden distribuir comercialmente dispositivos médicos. Se aprobó una nueva norma para incorporar la norma ISO 13485:2016 Dispositivos médicos - Sistemas de gestión de calidad dentro del QSR en un esfuerzo por armonizar los requisitos en todos los ámbitos. Muchas otras autoridades reguladoras de todo el mundo utilizan la norma ISO 13485:2016 como estándar para los sistemas de gestión de calidad. Si bien la flexibilidad de la autorización 510(k) permite a los fabricantes colocar su dispositivo en el mercado, la FDA puede inspeccionarlo en cualquier momento. Por lo tanto, lo mejor para el fabricante es garantizar que exista un sistema de gestión de calidad compatible antes de comercializar el dispositivo.
Con EU MDR, ISO 13485:2016 no es un requisito para el marcado CE. El Sistema de Gestión de la Calidad implantado deberá cumplir con las normas establecidas en el Anexo IX. Los fabricantes deberán cumplimentar una solicitud de evaluación de su Sistema de Gestión de Calidad ante el Organismo Notificado. Además de una auditoría del Sistema de Gestión de Calidad, el Organismo Notificado evaluará la documentación técnica de los dispositivos comparándola con el Sistema de Gestión de Calidad para garantizar que se cumplan todos los requisitos antes de la certificación.
Para armonizar el Sistema de Gestión de Calidad, la mejor práctica es implementar un sistema compatible con ISO 13485:2016 y el Programa de Auditoría Única de Dispositivos Médicos (MDSAP) con requisitos de cumplimiento adicionales para la FDA, EU MDR, Health Canada u otros países donde el fabricante pretende hacer negocios. .
Documentación
La FDA y el MDR de la UE exigen un proceso de diseño controlado para los dispositivos.
La presentación 510(k) requiere un Archivo de Historial de Diseño (DHF) completo que detalla los requisitos del sistema del dispositivo, la arquitectura, las especificaciones, la verificación y validación y la documentación de las actividades de gestión de riesgos. El perfil 510(k) eSTAR permite a los fabricantes adjuntar el documento relacionado a cada sección de la solicitud. La FDA proporciona una Lista de verificación de aceptación para guiar al fabricante con el proceso 510(k) y garantizar que exista la documentación correcta.
Para EU MDR, un Archivo Técnico contiene los documentos DHF y también requiere que el fabricante proporcione las siguientes listas de verificación adicionales:
- Requisitos generales de seguridad y rendimiento (GSPR) según el Anexo I, anteriormente conocidos como Lista de verificación de requisitos esenciales según MDD
- Documentación técnica estándar (SteD) según el anexo II
- Vigilancia poscomercialización (PMS) según el anexo III
Un documento técnico de alto nivel generalmente hace referencia a los diversos documentos y listas de verificación del DHF.
En ambos casos, se requiere un archivo completo de gestión de riesgos que cumpla con la norma ISO 14971.
Referencias:
- Notificación previa a la comercialización 510(k) | FDA
- El programa 510(k): Evaluación de la equivalencia sustancial en las notificaciones previas a la comercialización [510(k)] (fda.gov)
- Decidir cuándo presentar un plan 510(k) para un cambio en un dispositivo existente: guía para el personal de la industria y de la Administración de Alimentos y Medicamentos (fda.gov)
- Decidir cuándo presentar un plan 510(k) para un cambio de software en un dispositivo existente: borrador de guía para el personal de la industria y la Administración de Alimentos y Medicamentos (fda.gov)
- MDCG 2019-15, Notas orientativas para fabricantes de dispositivos médicos de Clase I
- Envíe y realice un seguimiento de los envíos previos a la comercialización de dispositivos médicos en línea: Portal CDRH | FDA
- Base de datos EUDAMED – EUDAMED (europa.eu)
- Enmiendas a las tarifas de usuario de dispositivos médicos (MDUFA) | FDA
- MDCG 2023-2, Lista de tarifas estándar
- Regulación del Sistema de Calidad (QS)/Buenas Prácticas de Fabricación de Dispositivos Médicos | FDA
- Listas de verificación de aceptación para 510(k)s | FDA
- Programa eSTAR | FDA
- FDA Health Canada eSTAR (starfishmedical.com)
Imágenes: Adobe Stock y estrella de mar médico
Dhruvitha Krishna es un Especialista en QA/RA en StarFish Medical con una maestría en Ingeniería Biomédica. Ha trabajado en manufactura, implementación de nuevos productos, implementación de software, gestión de proyectos y áreas regulatorias en empresas de dispositivos médicos. Dhruvitha se dedica a la mejora continua, regulatoria y de calidad de los procesos para el fabricante.
- Distribución de relaciones públicas y contenido potenciado por SEO. Consiga amplificado hoy.
- PlatoData.Network Vertical Generativo Ai. Empodérate. Accede Aquí.
- PlatoAiStream. Inteligencia Web3. Conocimiento amplificado. Accede Aquí.
- PlatoESG. Carbón, tecnología limpia, Energía, Ambiente, Solar, Gestión de residuos. Accede Aquí.
- PlatoSalud. Inteligencia en Biotecnología y Ensayos Clínicos. Accede Aquí.
- Fuente: https://starfishmedical.com/blog/medical-device-510k-ce-marking/

